
FEDERAL COURT OF AUSTRALIA
Peterson v Merck Sharpe & Dohme (Aust) Pty Ltd [2010] FCA 180
|
Citation: |
Peterson v Merck Sharpe & Dohme (Aust) Pty Ltd [2010] FCA 180 |
|
|
Parties: |
||
|
File number: |
VID 451 of 2006 |
|
|
Judge: |
JESSUP J |
|
|
Date of judgment: |
5 March 2010 |
|
|
Corrigenda: |
18 June 2010 |
|
|
Catchwords: |
TORTS – Negligence – Product Liability – Prescription medicine for relief of inflammation – Side-effects – Clinical trial presenting signal of cardiovascular risk – Whether discharge of duty of care required medicine to be withdrawn from market – Whether doctors, pharmacists, health care professionals and the public should have been warned – Whether they were warned – Terms of warning that would have been sufficient – Whether sufficiently communicated to doctors – Whether amendment to “Product Information” was sufficient – Manufacturer’s sales representatives claiming medicine safe – Whether justified claim – Whether duty of care breached by failing to warn and/or making safety claim. TORTS – Negligence – Product Liability – Causation – Prescription medicine for relief of inflammation contributing to applicant’s heart attack – Manufacturer negligently failed to warn of risk and represented safety of medicine – Whether applicant would have declined to take medicine if appropriately warned or if no safety claim made – Whether applicant’s doctor would have chosen not to prescribe medicine. TRADE PRACTICES – Misleading or deceptive conduct – Prescription medicine for relief of inflammation associated with doubling of risk of heart attack – No warning provided to doctors – “Product Information” for medicine not referring to increased risk – Whether misleading or deceptive conduct on part of corporation marketing the medicine – Sales representatives of corporation claiming medicine safe – Whether misleading or deceptive – Medicine contributed to applicant’s heart attack – Whether applicant’s doctor relied on conduct of corporation – Whether he would have prescribed medicine in any event. TRADE PRACTICES – Defective goods – Prescription medicine for relief of inflammation associated with doubling of risk of heart attack – No warning provided to doctors – Whether safety of medicine not as such persons generally entitled to expect – Whether defect arose only because of compliance with mandatory standard – Whether state of scientific knowledge not such as to enable defect to be discovered. TRADE PRACTICES – Unsuitable goods – Prescription medicine for relief of inflammation associated with doubling of risk of heart attack – No warning provided to doctors – Whether applicant made purpose of acquiring medicine known to manufacturer – Whether medicine not reasonably fit for that purpose – Whether applicant relied on manufacturer’s skill or judgment. TRADE PRACTICES – Goods of unmerchantable quality – Prescription medicine for relief of inflammation associated with doubling of risk of heart attack – No warning provided to doctors – Whether medicine not as fit for the purpose for which goods of that kind commonly bought as was reasonable to expect. |
|
|
Legislation: |
Federal Court of Australia Act 1976 (Cth) Pt IVA Therapeutic Goods Act 1989 (Cth) ss 4, 9D, 20, 23, 25, 28, 29A, 31, 32, 42DL Trade Practices Act 1974 (Cth) ss 4, 4C, 52, 74B, 74D, 75AA, 75AC, 75AD, 75AK, 82 |
|
|
Cases cited: |
Bowling v Pfizer, Inc 143 FRD 141 (1992) Briginshaw v Briginshaw (1938) 60 CLR 336 Carey-Hazell v Getz Bros & Co (Aust) Pty Ltd [2004] FCA 853 Chugg v Pacific Dunlop Ltd (1990) 170 CLR 249 Concrete Constructions (NSW) Pty Ltd v Nelson (1990) 169 CLR 594 Demagogue Pty Ltd v Ramensky (1992) 39 FCR 31 Donoghue v Stevenson [1932] AC 562 Dovuro Pty Ltd v Wilkins (2003) 215 CLR 317 E v Australian Red Cross Society (1991) 27 FCR 310 E v Australian Red Cross Society (1991) 31 FCR 299 Ferdinands v Commissioner for Public Employment (2006) 225 CLR 130 Frost v Aylesbury Dairy Company Ltd [1905] 1 KB 608 Graham Barclay Oysters Pty Ltd v Ryan (2002) 211 CLR 540 Grant v Australian Knitting Mills Ltd [1936] AC 85 Holman v Deol [1979] 1 NSWLR 640 Johnson Tiles Pty Ltd v Esso Australia Pty Ltd (2000) 104 FCR 564 Malec v JC Hutton Pty Ltd (1990) 169 CLR 638 Medtel Pty Ltd v Courtney (2003) 130 FCR 182 R v Small Claims Tribunal; ex parte Gibson [1973] Qd R 490 Rasell v Cavalier Marketing (Australia) Pty Ltd [1991] 2 Qd R 323 Reyes v Wyeth Laboratories 498 F 2d 1264 (1974) Rogers v Whitaker (1992) 175 CLR 479 Rosenberg v Percival (2001) 205 CLR 434 Ryan v Great Lakes Council [1999] FCA 177 Seltsam Pty Ltd v McGuiness (2000) 49 NSWLR 262 Sibley v Kais (1967) 118 CLR 424 Thompson v Johnson and Johnson Pty Ltd [1991] 2 VR 449 Vines v Djordjevitch (1955) 91 CLR 512 Wright v Dunlop Rubber Co Ltd (1972) 13 KIR 255 Wyong Shire Council v Shirt (1980) 146 CLR 40 |
|
|
|
|
|
|
Date of hearing: |
30, 31 March 2009 1-2, 6-9, 15-17, 21-24, 28, 29 April 2009 4-8, 11-15, 18-22, 25, 26 May, 2009 and 2, 9, 10-12, 22-25 June 2009 |
|
|
|
|
|
|
Place: |
Melbourne |
|
|
|
|
|
|
Division: |
GENERAL DIVISION |
|
|
|
|
|
|
Category: |
Catchwords |
|
|
|
|
|
|
Number of paragraphs: |
1014 |
|
|
|
|
|
|
Counsel for the Applicant: |
Mr J W K Burnside QC with Mr B F Quinn |
|
|
|
|
|
|
Solicitor for the Applicant: |
Slater & Gordon |
|
|
|
|
|
|
Counsel for the Respondents: |
Mr P R Garling SC with Mr G Williams |
|
|
|
|
|
|
Solicitor for the Respondents: |
Clayton Utz |
|
FEDERAL COURT OF AUSTRALIA
Peterson v Merck Sharpe & Dohme (Aust) Pty Ltd [2010] FCA 180
CORRIGENDA
1 Mr P Cashman also appeared as Counsel for the applicant.
2 At the start of para 368, "What Prof Celermajer described, “the IC50 curve”" should be "What Prof Celermajer described as “the IC50 curve”".
3 In Table 1 in para 405, the number “49” in the second column of the second row should be “99”.
4 In the final sentence of para 824, “warranty” should be “warning”.
5 In para 891, “see para 9 above” should read “see para 883 above”.
|
I certify that the preceding five (5) numbered paragraphs are a true copy of the Corrigendum to the Reasons for Judgment herein of the Honourable Justice Jessup. |
Associate:
Dated: 18 June 2010
|
IN THE FEDERAL COURT OF AUSTRALIA |
|
|
|
VICTORIA DISTRICT REGISTRY |
|
|
|
GENERAL DIVISION |
VID 451 of 2006 |
|
|
BETWEEN: |
GRAEME ROBERT PETERSON Applicant
|
|
|
AND: |
MERCK SHARPE & DOHME (AUSTRALIA) PTY LTD (ACN 000 173 508) First Respondent
MERCK & CO, INC. Second Respondent
|
|
|
JUDGE: |
JESSUP J |
|
|
DATE OF ORDER: |
5 MARCH 2010 |
|
|
WHERE MADE: |
MELBOURNE |
|
THE COURT ORDERS THAT:
1. The proceeding be listed for the making of orders on a date to be fixed, not before 6 April 2010.
2. Not less than 7 days before the date so fixed:
(a) the applicant file and serve a minute of the orders proposed by him to reflect the court’s determination of his personal claim;
(b) the second respondent file and serve a minute of the orders proposed by it to reflect the court’s determination of the applicant’s claim in negligence against it;
(c) the parties exchange, and file with the court, minutes of their respective proposals as to the orders which should be made pursuant to the schedule to the order made under s 33ZF of the Federal Court of Australia Act 1976 (Cth) on 30 March 2009; and
(d) any party who or which seeks the making of any other order at this stage of the proceeding, including any order as to costs, provide written notice thereof to the other parties.
Note:Settlement and entry of orders is dealt with in Order 36 of the Federal Court Rules.
The text of entered orders can be located using Federal Law Search on the Court’s website.
|
IN THE FEDERAL COURT OF AUSTRALIA |
|
|
VICTORIA DISTRICT REGISTRY |
|
|
GENERAL DIVISION |
VID 451 of 2006 |
|
BETWEEN: |
GRAEME ROBERT PETERSON Applicant
|
|
AND: |
MERCK SHARPE & DOHME (AUSTRALIA) PTY LTD (ACN 000 173 508) First Respondent
MERCK & CO, INC. Second Respondent
|
|
JUDGE: |
JESSUP J |
|
DATE: |
5 MARCH 2010 |
|
PLACE: |
MELBOURNE |
REASONS FOR JUDGMENT
table of contents
|
|
Commencing at |
||
|
part 1 – introduction ........................................................................................ |
[1] |
|
|
|
part ii − The story of Vioxx in the USA...................................................... |
[22] |
|
|
|
Some conventions........................................................................................................... |
[22] |
|
|
|
Early studies of Vioxx..................................................................................................... |
[30] |
|
|
|
Protocol 023.................................................................................................................. |
[43] |
|
|
|
The Board of Scientific Advisers’ Meeting....................................................................... |
[48] |
|
|
|
The CV SOP................................................................................................................. |
[56] |
|
|
|
The US New Drug Application for Vioxx........................................................................ |
[63] |
|
|
|
Dr Oates’ correspondence.............................................................................................. |
[68] |
|
|
|
The VIGOR trial............................................................................................................. |
[72] |
|
|
|
The preliminary results of VIGOR................................................................................... |
[79] |
|
|
|
The significance of Protocol 061..................................................................................... |
[90] |
|
|
|
Notification of VIGOR results to Merck subsidiaries....................................................... |
[97] |
|
|
|
The 14‑day report to the FDA........................................................................................ |
[103] |
|
|
|
Ongoing consideration of VIGOR results......................................................................... |
[114] |
|
|
|
The VIGOR Clinical Study Report.................................................................................. |
[121] |
|
|
|
The Safety Update Report of 13 October 2000.............................................................. |
[131] |
|
|
|
The Merck pooled analyses............................................................................................ |
[143] |
|
|
|
The first pooled analysis.................................................................................................. |
[147] |
|
|
|
The Arthritis Advisory Committee meeting....................................................................... |
[151] |
|
|
|
The Konstam article........................................................................................................ |
[158] |
|
|
|
The second pooled analysis............................................................................................. |
[163] |
|
|
|
The FDA warning letter.................................................................................................. |
[168] |
|
|
|
The Reicin article............................................................................................................ |
[170] |
|
|
|
Development of a cardiovascular outcomes study............................................................ |
[172] |
|
|
|
The third pooled analysis................................................................................................. |
[178] |
|
|
|
The Weir article.............................................................................................................. |
[184] |
|
|
|
The fourth pooled analysis............................................................................................... |
[186] |
|
|
|
Other studies referred to by Dr Reicin............................................................................. |
[189] |
|
|
|
Protocol 203.................................................................................................................. |
[191] |
|
|
|
The cardiovascular results of APPROVe and the withdrawal of Vioxx............................. |
[193] |
|
|
|
part iii − vioxx in australia............................................................................ |
[200] |
|
|
|
The statutory and regulatory framework.......................................................................... |
[200] |
|
|
|
Registration of Vioxx on the ARTG................................................................................. |
[211] |
|
|
|
Introduction of Vioxx to the Australian Market................................................................ |
[226] |
|
|
|
Amendment to the Vioxx Product Information................................................................. |
[230] |
|
|
|
The Marketing of Vioxx in Australia................................................................................ |
[259] |
|
|
|
part iv − vioxx and the risk of cardiovascular disease............. |
[297] |
|
|
|
Introduction.................................................................................................................... |
[297] |
|
|
|
Overview of expert opinion............................................................................................. |
[304] |
|
|
|
The Bradford Hill criteria................................................................................................ |
[321] |
|
|
|
APPROVe..................................................................................................................... |
[327] |
|
|
|
The generalisability of the APPROVe results................................................................... |
[343] |
|
|
|
VIGOR and the role of naproxen.................................................................................... |
[348] |
|
|
|
The Merck arthritis studies.............................................................................................. |
[388] |
|
|
|
The Merck Alzheimer’s disease studies........................................................................... |
[400] |
|
|
|
VICTOR........................................................................................................................ |
[423] |
|
|
|
Other meta analyses........................................................................................................ |
[424] |
|
|
|
Other studies.................................................................................................................. |
[440] |
|
|
|
The mortality data........................................................................................................... |
[442] |
|
|
|
The dose relationship...................................................................................................... |
[459] |
|
|
|
Consistency of association across studies........................................................................ |
[464] |
|
|
|
The FitzGerald hypothesis............................................................................................... |
[477] |
|
|
|
Other aspects of a biological explanation......................................................................... |
[531] |
|
|
|
Susceptibility of the atherosclerotic vasculature................................................................ |
[551] |
|
|
|
Other Bradford Hill criteria............................................................................................. |
[563] |
|
|
|
Other evidence of risk relied upon by the applicant.......................................................... |
[565] |
|
|
|
Vioxx and the risk of myocardial infarction...................................................................... |
[567] |
|
|
|
Vioxx and the risk of thrombotic stroke........................................................................... |
[573] |
|
|
|
Vioxx and the risk of the other pleaded cardiovascular conditions.................................... |
[584] |
|
|
|
part v − what the respondents knew or ought to have known.......................................................................................................................... |
[585] |
|
|
|
Before the results of VIGOR were available.................................................................... |
[588] |
|
|
|
Merck’s interpretation of the VIGOR results................................................................... |
[593] |
|
|
|
From VIGOR to 31 December 2000.............................................................................. |
[615] |
|
|
|
2001.............................................................................................................................. |
[635] |
|
|
|
2002.............................................................................................................................. |
[663] |
|
|
|
2003.............................................................................................................................. |
[669] |
|
|
|
2004.............................................................................................................................. |
[675] |
|
|
|
Summary........................................................................................................................ |
[679] |
|
|
|
part Vi − the applicant’s own circumstances..................................... |
[683] |
|
|
|
The applicant.................................................................................................................. |
[683] |
|
|
|
The applicant’s back pain............................................................................................... |
[695] |
|
|
|
Dr Dickman.................................................................................................................... |
[699] |
|
|
|
The applicant’s experience with Vioxx............................................................................. |
[727] |
|
|
|
Continuity of the applicant’s use of Vioxx........................................................................ |
[742] |
|
|
|
The applicant’s heart attack............................................................................................ |
[759] |
|
|
|
Did Vioxx cause or contribute to the applicant’s heart attack?.......................................... |
[764] |
|
|
|
Consequences of the applicant’s heart attack................................................................... |
[774] |
|
|
|
part vii − the applicant’s case in negligence...................................... |
[782] |
|
|
|
The existence and content of a duty of care..................................................................... |
[782] |
|
|
|
Discharge of the duty of care........................................................................................... |
[788] |
|
|
|
Failure to warn............................................................................................................... |
[815] |
|
|
|
MSDA’s marketing representations................................................................................. |
[854] |
|
|
|
Causation....................................................................................................................... |
[861] |
|
|
|
part viii − the applicant’s case under the trade practices act.................................................................................................................................. |
[875] |
|
|
|
Implied repeal................................................................................................................. |
[875] |
|
|
|
Section 52...................................................................................................................... |
[882] |
|
|
|
Section 75AD................................................................................................................ |
[912] |
|
|
|
Section 74B................................................................................................................... |
[931] |
|
|
|
Section 74D................................................................................................................... |
[969] |
|
|
|
Compensation................................................................................................................ |
[985] |
|
|
Part i − introduction
1 This proceeding, which was transferred to the court pursuant to an order made by the Supreme Court of Victoria on 10 April 2006, is a representative one under Pt IVA of the Federal Court of Australia Act 1976 (Cth) (“the Federal Court Act”). The second respondent, Merck and Co, Inc. (“Merck”), is a corporation based in the United States of America engaged in the development, manufacture, distribution and sale of pharmaceutical products both in that country and, through its international subsidiaries, in many parts of the world, including Australia. The first respondent, Merck Sharpe & Dohme Australia Pty Ltd (“MSDA”) is one of those subsidiaries.
2 In the 1990s, scientists employed by Merck at Merck Research Laboratories (“MRL”) developed a new non‑steroidal anti‑inflammatory drug which came to be called “rofecoxib”. It was to be used in the treatment of arthritis, and was a member of a new class of drugs then being developed by a number of pharmaceutical companies which were said to provide relief from the inflammation and pain associated with arthritis without gastrointestinal side‑effects. In due course, Merck manufactured rofecoxib on a commercial scale and incorporated it into a new pharmaceutical product marketed as “Vioxx”. So far as the Australian situation was concerned, Merck supplied rofecoxib to MSDA, which manufactured the medicine which was marketed in this country.
3 Vioxx was first registered on the Australian Register of Therapeutic Goods (“the ARTG”) on 17 January 2000. From at least February 2001, MSDA distributed and sold Vioxx in Australia until it was removed from the market on 30 September 2004. The events which are controversial in this proceeding relate directly to the circumstances which gave rise to the world‑wide withdrawal of Vioxx on that date.
4 The applicant, Graham Robert Peterson, had suffered from back pain for many years when he was first prescribed Vioxx by his doctor, Dr John Dickman, on 10 May 2001. He found that Vioxx provided relief from pain without the adverse gastrointestinal side-effects which he had encountered with other drugs. He continued to take Vioxx regularly until it was withdrawn from the market. In December 2003, he had a serious heart attack, received prompt, and, it seems, effective, medical treatment, and made an uncomplicated recovery. At the time, it occurred neither to him nor to Dr Dickman that the heart attack was related to the consumption of Vioxx. But the withdrawal of Vioxx from the market in September 2004 – because of the occurrence of adverse cardiovascular experiences in a major clinical trial – gave the applicant cause to make that connection. In this proceeding, he alleges that the consumption of Vioxx caused or contributed to his heart attack, and that the respondents failed to discharge their duty of care to him in relevant respects. He also makes certain allegations against MSDA under the Trade Practices Act 1974 (Cth) (“the Trade Practices Act”), to which I shall come. He makes the same allegations on behalf of the other members of the group constituted under Pt IVA of the Federal Court Act.
5 The proceeding is concerned with the operation in vivo of a bifunctional enzyme which is ubiquitous in human cells, prostaglandin H2 synthase, also called cyclooxygenase (commonly, and hereinafter, abbreviated as “COX”). By its cyclooxygenase activity, the enzyme catalyses the conversion of arachidonic acid to prostaglandin G2 (PGG2), and then by its peroxidise activity, the enzyme catalyses the conversion of PGG2 to prostaglandin H2 (PGH2). Other enzymes then convert PGH2 to eicosanoids, 20‑carbon molecules with functions described in the 5th edition of Lehninger: Principles of Biochemistry (2008), by DL Nelson and MM Cox, at p 358, as follows:
Eicosanoids are paracrine hormones, substances that act only on cells near the point of hormone synthesis instead of being transported in the blood to act on cells in other tissues or organs. These fatty acid derivatives have a variety of dramatic effects on vertebrate tissues. They are involved in reproductive function; in the inflammation, fever and pain associated with injury or disease; in the formation of blood clots and the regulation of blood pressure; in gastric acid secretion; and in various other processes important in human health or disease … There are three classes of eicosanoids: prostaglandins, thromboxanes, and leukotrienes.
We are presently concerned only with the first and second classes of eicosanoids mentioned above: prostaglandins and thromboxanes.
6 There are three features of these biological processes that assume particular importance in the present case. First, although COX is ubiquitous, and although the sequence by which eicosanoids are derived from arachidonic acid is the same in all cells, the resulting hormones differ as between cell types. In the words of Prof Cleland, a rheumatologist called by the applicant: “While COX is ubiquitous, tissues vary in expression of terminal synthases …” Secondly, as stated in the extract from Lehninger, the eicosanoids “act only on cells near the point of hormone synthesis”. And thirdly, depending on location in the body, the action of COX may be constitutive (that is, operating all the time) or inducible (that is, operating only in response to particular stimuli).
7 Our present concern involves the functions of eicosanoids in three distinct parts of the body: the sites of arthritic joints, the stomach and upper duodenum, and the vasculature. In the vicinity of arthritic joints, COX is induced by the pathological state of the joint, but, in a situation in which the condition is chronic, the action of the enzyme is effectively continuous. The eicosanoids produced in this situation are the prostaglandins: most conspicuously prostaglandin E2 (PGE2), but also possibly, it seems, prostaglandin I2 (PGI2 – also known, and to which I shall refer, as prostacyclin). They are responsible for the inflammation which is the cause of much of the pain and inconvenience of arthritis. In the stomach and upper duodenum, COX operates constitutively to produce PGE2 (and possibly also prostacyclin) which contribute to the protection of the mucosal lining. In the words of Prof Yeomans, a gastroenterologist called by the respondents, the prostaglandins are “one of the important defence factors that help the stomach and duodenum resist being digested by their own corrosive, acid juice”. In the vasculature, COX operates at two levels which are presently relevant: in blood platelets, it is involved in the production of thromboxane A2 (TXA2 – to which I shall refer as thromboxane), a clotting and vaso‑constricting agent; and in the endothelium of blood vessels, it is involved in the production of prostacyclin, in that context an anti‑coagulant (by binding to a receptor on the surface of, and thereby rendering inactive, platelets that would otherwise tend to coagulate) and vaso‑dilating agent.
8 For many years non‑steroidal anti‑inflammatory drugs (“NSAIDs”) (including aspirin) have been used in the mitigation of the inflammation associated with arthritis. In 1971 it was discovered that the inhibition of the operation of COX was responsible for the analgesic and anti‑inflammatory effects of aspirin and other NSAIDs. This also provided an explanation for the gastrointestinal side-effects that were associated with the ingestion of NSAIDs: if the operation of COX were inhibited, the beneficial effect of the prostaglandins in the stomach and duodenum would likewise be compromised. Then in 1990 it was discovered that there were in fact two isoforms of COX, one which was constitutive, which came to be called COX-1, and another which was inducible, which came to be called COX-2. (It seems that, in the kidney and the brain only, COX-2 is to an extent constitutive, but otherwise the foregoing generalisation will be sufficient for present purposes.) It was discovered that the form which played a role in the stomach and duodenum was COX‑1, while the form which was involved in the inflammatory process in the joints was COX-2. This led to sustained efforts to develop compounds that would inhibit the operation of COX-2 only, leaving COX-1 untouched to continue its beneficial work in the gut. Before long, such compounds were developed, first in the form of a molecule called “celecoxib” which later became the active ingredient in a drug marketed as “Celebrex”, and shortly thereafter in the form of a molecule called “rofecoxib” which later became the active ingredient in Vioxx. These and other molecules which inhibit only COX-2 are referred to as COX‑2 inhibitors, or simply as “coxibs”.
9 The controversy in the present case centres around the effect which the consumption of coxibs, and of Vioxx in particular, has in the context of the vasculature. The applicant’s case relies on the circumstance that in platelets the production of thromboxane is the work of COX-1 (which is uncontroversial), while in the endothelium the production of prostacyclin is the work of COX-2 (which is not uncontroversial, but the applicant so alleges). He therefore contends, in short, that, unlike the traditional NSAIDs which act indiscriminately, Vioxx blocks only the production of endothelial prostacyclin in blood vessels, while leaving the production of platelet thromboxane unaffected. Without the mitigating effect of prostacyclin, it might be expected that the aggregation of platelets and the consequent clotting of the blood will proceed unrestrained, with thrombotic and/or thromboembolic consequences. The applicant contends that this is exactly what happened to him in December 2003, when he had his heart attack.
10 That brings me to the applicant’s case in court. The group members to whom the proceeding relates are identified in para 2 of the Further Amended Statement of Claim (“the Statement of Claim”) as follows.
The group members to whom this proceeding relates … are all persons who:
(a) after 30 June 1999 obtained from a medical practitioner in Australia one or more prescriptions of the non‑steroidal anti‑inflammatory drug rofecoxib sold as tablets under the trade mark or brand “Vioxx” … as pleaded in paragraph 6 herein; and
(b) after 30 June 1999 completed one or more prescriptions of Vioxx tablets purchased in Australia as pleaded in paragraph 7 herein; and
(c) at any time after completing their first prescription of Vioxx tablets purchased in Australia but before the day 30 weeks after last consuming a Vioxx tablet, suffered and were diagnosed as having suffered one or more of the following conditions:
(i) myocardial infarction;
(ii) thrombotic stroke;
(iii) unstable angina;
(iv) transient ischaemic attack;
(v) peripheral vascular disease; …
I shall refer to these conditions as “the pleaded cardiovascular conditions”.
11 It is alleged that, after 30 June 1999, each of the group members obtained from a medical practitioner in Australia one or more prescriptions of Vioxx, and subsequently “completed” those prescriptions by taking Vioxx tablets purchased in Australia. It is alleged that, after completing his or her first prescription of Vioxx tablets, but before the end of the 30th week after last consuming a Vioxx tablet, each of the group members suffered one or more of the pleaded cardiovascular conditions. In the case of the applicant, it is alleged, and it is common ground, that he suffered a myocardial infarction on 8 December 2003. It is alleged that each pleaded cardiovascular condition suffered by each group member was caused by his or her consumption of Vioxx tablets, and that he or she experienced pain or suffering, and suffered loss and damage, as a consequence.
12 The applicant’s cause of action against Merck, and a significant cause of action against MSDA, sounds in negligence. He alleges that the consumption of rofecoxib “materially increased the risk” of suffering the pleaded cardiovascular conditions. He alleges that the respondents “knew or ought to have known” of that circumstance. He alleges that the respondents owed to each of the group members a duty to take reasonable care to avoid acts and omissions that may have exposed them to a material increase in the risk of suffering the pleaded cardiovascular conditions as a consequence of consuming Vioxx tablets. He alleges that the respondents failed to undertake the necessary research, or to have regard to the results of their research, as to the side‑effects and health risks associated with Vioxx, that they continued to market Vioxx rather than awaiting the results of such research as was being undertaken and, in the case of MSDA, that it failed to inquire of Merck about the possible side‑effects and health risks, and manufactured Vioxx from rofecoxib rather than from some other drug.
13 The applicant alleges against MSDA that it failed to warn medical practitioners, pharmacists, other health care professionals and consumers of the risks said to be associated with the consumption of Vioxx. He says that such warnings ought to have been given in various places and by various means, to which I shall turn. In these respects, he adds an allegation that Merck too failed to warn.
14 The applicant alleges that the respondents failed until 30 September 2004 to withdraw or to recall Vioxx tablets from the market. These allegations are denied by the respondents, although it is common ground that they did not withdraw or recall Vioxx tablets until 30 September 2004.
15 The applicant also advances a case of commission against MSDA, alleging that it undertook a sophisticated marketing campaign for Vioxx, by which the overall safety of the drug was emphasised in written and oral communications with medical practitioners and others, and by which such evidence as there was of the cardiovascular risks associated with the consumption of Vioxx was de‑emphasised at best, and often either wilfully or carelessly misrepresented. These allegations are used by the applicant both in his negligence case (in the sense that, notwithstanding the cardiovascular risk said to be associated with the consumption of Vioxx, and the absence of adequate research or investigation to resolve that risk, MSDA deployed these representations with a view to securing the greater use of Vioxx in Australia) and in his case under the Trade Practices Act, in respects to which I shall turn in due course.
16 By reason of the matters to which I have referred, the applicant alleges that the respondents failed to take reasonable care to avoid acts and omissions that may expose the group members to a material increase in the risk of suffering the pleaded cardiovascular conditions as a consequence of consuming Vioxx tablets. In the case of Merck, this breach of duty is alleged to relate to the manufacture, and supply to MSDA, of rofecoxib. In the case of MSDA, the breach of duty is alleged to relate to its manufacture, packaging, labelling, marketing, distribution and supply for sale of Vioxx tablets. In each case, it is said that the breach of duty caused each of the group members to be prescribed Vioxx, to complete one or more prescriptions of Vioxx tablets, to suffer one or more of the pleaded cardiovascular conditions, to experience pain and suffering, and to suffer loss and damage.
17 The applicant also proceeds against MSDA under ss 52, 75AD, 74B and 74D of the Trade Practices Act. He alleges that MSDA’s failure to provide any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the pleaded cardiovascular conditions was conduct which occurred in trade and commerce, and was misleading or deceptive, or likely to mislead or deceive, in contravention of s 52. The applicant relies also on the representations to which I have referred to in para 15 above, alleging that those representations were misleading or deceptive, or likely to mislead or deceive, because the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the pleaded cardiovascular conditions, and because Vioxx was not safer in that respect than comparable drugs. Under s 75AD, the applicant says that Vioxx tablets were goods manufactured by MSDA, that the goods had a defect and that the group members suffered injuries because of the defect. To establish that Vioxx tablets had a “defect” in the relevant sense, the applicant invokes s 75AC, alleging that the safety of Vioxx tablets was not such as persons generally were entitled to expect. Under s 74B, the applicant alleges that Vioxx tablets were not reasonably fit for the purpose for which they were, expressly or by implication, acquired by consumers, that they were supplied by MSDA as manufacturer to medical practitioners and pharmacists in trade and commerce, and that such persons acquired the goods for the purpose of re‑supply to consumers, including the group members. The applicant makes like allegations that Vioxx tablets were not of merchantable quality, and thus supplied by MSDA in contravention of s 74D.
18 There was virtually no aspect of the applicant’s case with which the respondents did not join issue. They deny that the consumption of Vioxx increased the risk of occurrence of any of the pleaded cardiovascular conditions. They say that the withdrawal of Vioxx from the market in September 2004 was a prudent and cautious expedient which in no sense implied that the consumption of Vioxx was harmful. Alternatively, they say that, at all times until then, they carefully and conscientiously monitored the emerging science as to the possibility of a causal link between rofecoxib and cardiovascular disease, and that, until September 2004, the existence of such a link was not reasonably apparent to them. They say that they did carry out reasonable research and investigations in this regard. They resist the proposition that they failed to advise doctors and others of such information about the possible link between rofecoxib and cardiovascular disease as was actually or constructively known to them. MSDA says that, by including what it knew (at various stages) about that subject in the “Product Information” maintained in connection with the registration of Vioxx on the ARTG, it took such reasonable steps as were required of it in this regard. It says that, because Vioxx was a prescription‑only medicine, it discharged its duty of care by addressing such advices, warnings and the like as were necessary from time to time to doctors, and that it was not necessary for it to have done so directly to patients, or to the public generally. MSDA says that the marketing campaign on which the applicant relies was, as a matter of policy and strategy, entirely internal to itself, and that the only facts which would assist the applicant, in any aspect of his case, were proven communications made to persons external to MSDA which had the potential to affect the prescribing decisions of doctors.
19 The respondents also rely on a number of specific defences – both at common law and under the Trade Practices Act – upon which it is not necessary to elaborate at this stage. Their Defence travelled a good deal further in this respect than their submissions in the case. To the extent necessary, I shall deal with the respondents’ defences as the context requires in my reasons below.
20 The circumstance that the present proceeding is a representative one under Pt IVA has made it necessary to deal with more issues than presently arise in the resolution of the applicant’s personal case. In due course the resolution of those issues will be linked with the claims of other group members as the applicant brings them forward. The extent to which I am presently obliged to determine those issues was the subject of an order made on 30 March 2009, a copy of which follows these reasons as Appendix A. That order was ultimately made by consent, but that it was made at all was the result of contested proceedings and of a judgment of the Full Court given on 11 March 2009: Merck Sharp & Dohme (Australia) Pty Ltd v Peterson [2009] FCAFC 26. At the conclusion of the trial, counsel for both parties proposed that I should leave the precise formulation of the terms in which answers might be given to the questions referred to in the order until they and their clients had had the opportunity to consider my reasons. I shall follow that course.
21 In these reasons, I propose to commence by tracing the development of Vioxx in the United States of America. It will be necessary to refer in some detail to the trials and studies of, or which were relevant to, Vioxx, including those on which the parties rely to implicate Vioxx in, or to exculpate it from, a causal involvement in the onset of thrombotic cardiovascular disease. The introduction of Vioxx into Australia, and the interactions between MSDA and the relevant regulator in that respect, will then be examined. I shall then consider whether Vioxx did, as an objective fact, have a tendency to give rise to cardiovascular conditions. If it did, it will then be necessary to consider whether, at some point before Vioxx was withdrawn in September 2004, the respondents knew or ought to have known of that tendency. I shall then consider the facts relating to the applicant himself, including the question whether Vioxx caused or contributed to his heart attack. From there I shall move to the applicant’s causes of action in negligence and under the Trade Practices Act. Although dealt with in the setting of the applicant’s own case, those parts of my reasons will necessarily include a number of determinations and findings of relevance to the group members as a whole, which will, therefore, be reflected in the answers which I shall, in due course, provide to at least a number of the questions set out in Appendix A.
part ii − The story of Vioxx in the USA
Some conventions
22 Before turning to the story of Vioxx as such, I should lay down certain conventional foundations for much of the discussion which will follow. First, it will be necessary to refer to many published articles in the scientific and medical literature. In order to avoid burdening these reasons with lengthy and often repetitive full citations, I have listed the articles to which I shall refer in Appendix B. In my reasons as such, I shall refer only to the name of the first‑mentioned author, followed by the year of publication, as, for example, Konstam (2001).
23 Secondly, as will become apparent, Vioxx and other like medicines were the subject of many trials and studies, both general and specific. It will be useful at this stage to identify the four main types of investigation that are employed by drug companies and medical researchers. For the most part, what follows is taken from the evidence of Dr Alise Reicin, a Vice President of Research and Early Development at MRL, although I have supplemented that with the understanding I have derived from the evidence in the case generally. Dr Reicin said that a clinical pharmacology study was one in which –
… you actually look at how the drug is absorbed or metabolised, the pharmacokinetics of the drug or its specific effects on enzymes or specific end products in the body. You want to look at very specific questions about how your drug affects specific products in the body.
Such a study is often a rather small one, in which the pharmacological effect of the drug is studied in a sample of volunteers, either healthy or, on occasions, selected for a characteristic of interest (such as a particular medical condition).
24 An observational study is usually, though not invariably, a retrospective study making use of a database of patients and their conditions. Dr Reicin agreed that it was in the nature of an ex post facto systematic examination of clinical or pathological outcomes in a range of cases which had happened in real life. Of such a study, she said:
So you will go into a hospital database and you will say, “Okay, how many patients had heart attacks?” Then you will say, “Of those patients who had heart attacks and those patients who didn’t have heart attacks what percentage were on this drug versus what percentage were on that drug?” and you will try and make observations and conclusions as to whether the percentage on X drug was higher in patients who had the event versus those who did not … The level of certainty around that observation is much less than what you would get in a randomised clinical trial.
25 The trial last referred to in the passage above is that which is regarded by the researchers as the “gold standard” of clinical trials: the prospective, randomised, double‑blind, placebo‑controlled or comparator‑controlled trial. In these reasons, I shall abbreviate this to randomised clinical trial (“RCT”). Such a trial is an “outcomes‑based” one because it proceeds by way of the enrolment of a number (ideally a large number) of patients, randomizes them to the drug or to the comparator in separate arms or groups, and records the clinical outcomes which they encounter over the study period (which may be the period of the trial as a whole, or it may be a predetermined time for which each patient should take his or her study therapy). Typically patients are enrolled for such a trial with the co‑operation of their own treating practitioners, who then report to the administrators of the trial what clinical outcomes the patients have encountered as required. The practitioners are, in such a role, referred to as “investigators”.
26 Finally (for present purposes) there is the meta‑analysis, or pooled analysis. This is an analysis of the results of other trials or studies, and may be either retrospective (the more common form) or prospective (as in the case of an important study in the history of Vioxx). Here there have been, or there will be, a number of other trials and studies, each with its own purposes, but more statistically useful information as to a particular outcome or condition may be derived by pooling the results of them. As I observed in a number of such instances dealt with in the evidence, a meta‑analysis may be valuable where the event of interest was not the primary outcome studied in the underlying trials and where, because of the small numbers of occurrences, the data from those trials do not, separately, sustain a meaningful conclusion as to association. In such cases, pooling the trial results may give the biostatistician the required numbers to take that extra step.
27 Thirdly, in what follows there will be repeated references to a concept known as the “relative risk”. A commonly used convention for the expression of the risk of the occurrence of a particular adverse event resulting from taking a medication of interest is to express the risk per 100 patient‑years in the trial or study concerned. For example, if there were 5000 patients in one arm of a trial, and they all remained in the trial for 18 months, the total patient-years would be 7500. If there were 50 adverse events in that arm, the risk would be represented as 50/(7500/100) = 0.67. One then comes to the concept of “relative risk”, which is the risk in one arm of the trial compared to the risk in the other. Reduced to its simplest essentials, in the example given, if the risk in the second arm were, say, 1.45, the relative risk of taking the medication in that arm would be 1.45/0.67 = 2.16. That figure, however, represents only the relative risk as between the actual recorded experiences in the trial. The real‑world relative risk of taking the second medication rather than the first is a matter of statistical estimation, based upon the size and other characteristics of the sample used in the trial. Here, by convention, statisticians identify a range, or interval, within which the true relative risk would be found 95 times out of 100 (ie it is the interval within which the relative risk would be found 95 times if the same study were conducted 100 times).
28 The single figure derived in the way explained above is, also by convention, generally stated, but this is no more than what has been described as the “point estimate”. The theoretical point at which there is no difference between the risks of taking the two medications is unity. If the 95% confidence interval straddles 1, one could not say, with this measure of confidence, that the risk of taking one medication is greater (or less) than the risk of taking the other. One could not say, by this conventional measure, that the difference in risk between the two medications was significant. Thus, although the point estimate of 2.16 in the example given above may appear to suggest that the risk of taking one medication is more than twice as great as that of taking the other, if the 95% interval were, say, 0.85 – 4.45, the statistician would not conclude that the difference was significant by reference to the convention to which I have referred. In the reasons which follow, I shall use the following conventional format for the expression of relative risk, and the 95% confidence intervals associated with that (taking by way of example the numbers used above): 2.16 (0.85, 4.45).
29 The other statistical concept often referred to in the evidence is the “p‑value”. As explained by one of the biostatisticians called by the applicant, Prof Woodward, this is a measure of the probability of being wrong in the rejection of a null hypothesis. In the context of relative risk, the null hypothesis conventionally used is that there is no difference between the two treatments (for instance) being compared. Put another way, the null hypothesis is that the relative risk of taking one treatment rather than the other is unity. Thus, although the 95% confidence interval of the relative risk in a particular case may be, say, 1.20 – 3.75, if the p-value were, say 0.01, one would conclude that the probability of the real relative risk being 1 (the null hypothesis which is presumptively rejected) would be 1 in 100. The p‑value thus provides additional information in cases in which the 95% confidence interval for the relative risk lies wholly above or wholly below unity. As I understand it, the p‑value may also provide information in cases in which there is no statistically significant difference at the 95% level. By definition, the probability of a rejection of the null hypothesis being wrong will always be greater than 0.05, but it may sometimes be of interest to know by how much it is greater.
Early studies of Vioxx
30 The development of Vioxx at MRL, and the clinical trials conducted in that context, were the subject of the evidence of Dr Reicin. She joined Merck in 1996 as an Associate Director in the Pulmonary/Immunology department of MRL, and, in early 1997, began to work on Vioxx. She was first involved in the clinical tests of Vioxx that would form the basis of Merck’s submission to the Food and Drug Administration (“the FDA”) for the purpose of the approval of Vioxx for prescription use in the United States. She subsequently worked extensively on Vioxx until after its voluntary withdrawal in September 2004.
31 Dr Reicin said that, in the United States, the FDA is the government agency that exclusively regulates the approval and marketing of new medicines. She said that prescription drugs go through a series of rigorous scientific tests and trials before they can be approved and marketed, including pre‑clinical trials (such as animal trials) and clinical (ie human) trials. During the pre‑clinical phase, new drugs are evaluated first in vitro, and are then tested in animals to determine whether they may be safely given to human subjects.
32 Before a new drug is tested in humans in the United States, a drug sponsor must submit an Investigational New Drug Application to the FDA. Such an application contains extensive information on the manufacture of the drug, as well as the data from pre‑clinical and other studies. Based on the information submitted in it, the FDA reviews the proposed protocols for trials in humans, and decides whether to allow those trials to proceed.
33 A new drug must undergo three phases of clinical trials before it is eligible for marketing approval in the United States. Phase I clinical trials involve the administration of the drug to a small number of healthy volunteers to test the drug’s safety and tolerability, its metabolic and pharmacologic actions, and the pharmacokinetics associated with increasing doses. Phase II clinical trials are designed to establish the effectiveness of the drug in the intended patient population, and to explore the appropriate dose range for the drug. Phase III clinical trials gather additional information about a drug’s safety, effectiveness, and dosage. They are generally the largest and longest‑term studies, and are often planned and conducted in consultation with the FDA.
34 When a sponsor believes that it has sufficient data to demonstrate that the drug is safe and effective if used as intended, the sponsor submits to the FDA a New Drug Application containing the data gathered during the pre‑clinical and clinical trials. That application also contains the sponsor’s proposed labelling for the drug. It contains all known information regarding the drug, including a summary of the efficacy and safety data, and integrated assessments of benefit and risk. On the basis of that application, the FDA will determine whether the drug is safe and effective, and also rule on the content, placement and language of the drug’s labelling.
35 Dr Reicin said that the drug that was to become Vioxx went through the stages referred to above. It was studied in test tubes and animals to examine its pharmacological properties, its efficacy, and its safety. Merck submitted the Investigational New Drug Application to the FDA on 16 December 1994. In the Phase I clinical trials, the effects of single oral doses ranging from 5 mg to 1000 mg in young healthy male volunteers were studied. Next, the effects of oral doses ranging from 25 mg to 375 mg over a two‑week period were studied. Studies were also undertaken with healthy older people, using doses up to 250 mg. Then in the Phase II clinical trials, the drug was tested to assess its efficacy in the intended populations – patients with chronic and acute pain. Specifically, the Phase II studies included two patient populations: patients with pain following dental surgery and patients with pain associated with osteoarthritis. Merck also studied the efficacy of the drug for patients with rheumatoid arthritis. All these studies showed that the drug was effective at relieving the pain and inflammation associated with the conditions mentioned. The Phase III clinical trials, which were planned in consultation with the FDA, were designed to assess whether the drug treated pain as well as existing drugs, but with a reduced incidence of gastrointestinal problems.
36 Although not mentioned in terms by Dr Reicin, there are aspects of clinical trials of which I was informed by counsel for the respondents in opening, and which appear to be uncontroversial. The Phase II trials are divided into two categories – IIa and IIb. Only the latter have played any part in the evidence in this proceeding. I was told that Phase IIa trials were concerned with the appropriate dose ranges for a drug: they do not need to be further mentioned. It was the Phase IIb efficacy trials of Vioxx that featured significantly in the evidence. I was also informed by counsel for the respondent that trials conducted after a drug is on the market are referred to as Phase IV trials, and that trials used to explore the prospect of a new indication are sometimes referred to as Phase V.
37 As part of the clinical development program for its new drug, Merck also conducted endoscopy studies, with a view to assessing, by endoscopic inspection, the occurrence of gastrointestinal mucosal damage or ulcers in patients taking the drug or a comparator. Those studies showed that the new drug, even when administered at very high doses, was associated with less gastroduodenal damage than was observed in subjects taking comparator NSAIDs. These results were consistent with the theoretical, and expected, action of a coxib, namely, that it had comparable efficacy to other NSAIDs in the relief of pain and inflammation, but was associated with lower levels of endoscopically visible gastrointestinal damage.
38 Dr Reicin said that, in the endoscopy studies just referred to, the majority of the ulcers observed were asymptomatic. She said that, in order to determine whether the reduction in asymptomatic ulcers would translate into a reduced incidence of the serious and potentially fatal gastrointestinal perforations, ulcers, and bleeds, it was necessary to examine the rates of these outcomes in clinical trials. Here a number of difficult methodological questions had to be resolved. Among them was the question whether to include patients who had been prescribed low‑dose aspirin by their doctors to prevent heart attacks. The reason why aspirin is such a widely‑used medication for the avoidance of thromboembolic events is its unique effect upon platelet COX-1. Aspirin irreversibly acetylates COX-1, thereby rendering the platelet inactive for the term of its life (approximately four days). The effect of other NSAIDs upon COX-1 is reversible (a subject to which I shall return). The problem with including patients who were taking aspirin in any proposed clinical trial was that the inhibition of COX-1 by aspirin could lead to the very gastrointestinal outcomes that MRL was trying to test. That is to say, by allowing the use of aspirin in the study, MRL would have compromised the integrity of its attempt to test the hypothesis that the new drug would be associated with fewer gastrointestinal bleeds, for example, than a non‑selective NSAID that inhibited both COX-1 and COX-2.
39 On the other hand, according to Dr Reicin, there were also good reasons for including patients who were taking low‑dose aspirin in the study. One was the reality that, in the real world, many people did take low‑dose aspirin. Another reason was based on the expectation that the comparator group would have a traditional NSAID administered to them. Dr Reicin was aware that such NSAIDs, because of their inhibition of COX-1, had anti‑clotting effects, at least to an extent, like aspirin. The group taking the new drug, because it did not inhibit COX-1, might therefore be expected to encounter a higher rate of cardiovascular adverse events. As to this concern, Dr Reicin referred to a memorandum by her colleague, Dr Thomas Musliner dated 21 November 1996. Amongst other things, Dr Musliner said:
If patients are allowed to use low‑dose aspirin in an MK‑966 vs. NSAID comparator design study, the published data suggest that the “background” PUBs [ie perforations, ulcers and bleeds] rate in the selective Cox‑2 inhibitor group can be anticipated to be ~1.5 fold higher than if aspirin use were not permitted. Since the GI [ie gastrointestinal] effects of low‑dose aspirin (particularly if an enteric‑coated formulation is used) are predominantly attributable to its antiplatelet effect (rather than any local effect) it is likely that GI risk will be increased more in the Cox‑2 inhibitor group than in the NSAID group, because platelet function will already be impaired in the latter group. …
If aspirin prophylaxis is not permitted, there is a substantial chance that significantly higher rates of CV AE events (MIs, angina, strokes, TIAs, etc.) will be observed in the selective Cox‑2 inhibitor group compared to the standard NSAID group … While one could argue that the differences are not unexpected due to the absence of antiplatelet effect for the selective Cox‑2 inhibitor, it would create a negative aspect to the results and leave open the question (reasonable or unreasonable) whether the drug might in some other way be contributing to such events.
What Dr Musliner referred to as “MK‑966” was the drug which later became Vioxx.
40 In February 1997, Dr Musliner sent Dr Reicin a draft of a gastrointestinal clinical outcomes protocol that he had helped prepare. It would compare MK‑966 with two non‑selective NSAIDs, diclofenac and ibuprofen. The draft proposed the exclusion of patients who needed to take low‑dose aspirin, but permitted “patients with a history of myocardial infarction or coronary arterial bypass grafting more than 1 year prior to study start” to participate. Dr Reicin said that patients such as these routinely received low‑dose aspirin for cardioprotection. She made a number of endorsements on the draft, including the following:
Do we want to allow high risk patients in the study given the risks we have discussed? I think this will definitely make enrollment more difficult, but I am still concerned that the NSAID group will be getting cardioprotection that the 966 group will not – I sent you and Alan an article on flurbiprofen given after PTCA – it decreased the reocclusion rate compared to placebo – no comparison to aspirin.
Dr Reicin referred to this endorsement to demonstrate that, at the time, the theory that she and her colleagues at MRL entertained was that the group in the study receiving the traditional NSAID would be getting cardioprotection, while the MK‑966 group would not. She said that she was not considering the possibility that MK‑966 might be prothrombotic.
41 The draft of the gastrointestinal outcomes protocol was sent also to Dr Briggs Morrison, another research scientist at MRL. He sent an email to Dr Reicin in which he said:
Would allow low dose aspirin – I know this has been discussed to death, but real world is everyone is on it, so why exclude AND without COX-1 inhibition you will get more thrombotic events and kill drug.
In a response to Dr Morrison, Dr Reicin wrote:
Low Dose Aspirin – I HEAR YOU! This is a no win situation! The relative risk of even low dose aspirin may be as high as 2‑4 fold. Yet, the possibility of increased CV [ie cardiovascular] events is of great concern – (I just can’t wait to be the one to present those results to senior management!). What about the idea of excluding high risk CV patients – ie those that have already had an MI, CABG, or PTCA? This may decrease the CV event rate so that a difference between the two groups would not be evident. The only problem would be – Would we be able to recruit any patients?
Dr Reicin was cross‑examined about this response. She resisted the suggestion that the parenthetical passage meant that she did not want to have to tell senior management that more cardiovascular events were being seen amongst patients taking Vioxx. She said:
I think it was more saying, “We would have done this large study. We would see a difference. There would be no placebo”. And while it was likely that the difference would be caused by the cardioprotection of NSAIDs, you couldn’t prove it, you had a new drug and you would have results that people would have a difficult time interpreting.
42 As events turned out, Merck did not proceed with the gastrointestinal outcomes study that had been the subject of Dr Musliner’s draft.
Protocol 023
43 A study which did proceed, and which was significant in the understanding of the mechanism of coxibs, was Protocal 023, a pharmacology study conducted between May 1996 and January 1997. According to Dr Reicin, this study was conducted because traditional NSAIDs were known to cause sodium and water retention by inhibiting the synthesis of prostaglandins in the kidney. It was known that both COX-1 and COX-2 were constitutively expressed in the kidney, but it was unclear whether these side-effects were caused by the inhibition of COX-l or of COX-2. Protocol 023 was designed to study whether patients receiving MK‑966 would experience salt retention similar to that experienced by patients receiving a traditional NSAID. In the study, 36 healthy older subjects were randomised to MK‑966, the traditional NSAID and placebo (12 per arm). They were confined for at least 17 days and placed on a special diet. Regular urine and blood samples were taken from them. The amount of salt in the urine was the primary measurement, but investigators also measured the urinary metabolites of prostacyclin and thromboxane, known as PGI‑M and TX‑M respectively.
44 Merck received the results of Protocol 023 some time after 29 August 1997. On 20 October 1997, Dr Morrison, the clinical monitor for the study, sent a memorandum to a number of his colleagues, including Dr Reicin. Relevantly to the present issue, Dr Morrison said:
The most surprising result from this study is the analysis of prostaglandin metabolism. In terms of systemic PG production, 2 things were analyzed. The first is … [a] metabolite of TxB2. … [T]he effect of MK‑0966 … was not different from placebo confirming COX-2 selectivity. The second is PGI‑M, which is a metabolite of prostacyclin which is largely derived from endothelial cells. Surprisingly, MK‑0966 had just as pronounced an effect on this endpoint as [the NSAID]. Although both COX-l and COX-2 are known to be expressed in endothelial cells, this result suggests that most of the prostacyclin production comes from COX-2. Alan and Barry are discussing this with Dr. Oates and other prostaglandin experts. Whether there is any clinical implication of inhibiting prostacyclin more than thromboxane is unclear.
According to Dr Reicin, MRL scientists reviewed the academic literature and conducted animal and human studies the better to understand the questions raised by the urinary metabolite data from Protocol 023.
45 Part of MRL’s consideration of the data from Protocol 023 was to undertake an analysis of the incidence of cardiovascular serious adverse events in those of its Phase IIb/III osteoarthritis clinical trials. The analysis was the subject of a report by Dr Doug Watson dated 2 February 1998. In the introductory section of his report, Dr Watson said:
In … Protocol 023 …, Vioxx (50 mg daily), and indomethacin (50 mg 3 times daily) were both found to significantly reduce the urinary excretion of PGI‑M compared to placebo. However, Vioxx had no effect on systemic thromboxane production (largely platelet derived) as assessed by measurements of serum TxB2 and the urinary metabolite, 11‑dehydro‑thromboxane B2. It was felt that the reduction in urinary PGI‑M likely reflects both a reduction in renal PGI2 generation … and to some undefinable extent a decrease in systemic PGI2 synthesis. … PGI2 is synthesized ubiquitously in blood vessel walls from arachidonate derived from either the vessel wall or the platelet. It is the most potent of all inhibitors of platelet aggregation, and has vascular dilatory properties as well. Conversely, TXA2, a potent platelet aggregator and vasoconstrictor, is synthesized primarily by the platelet … The clinical implications of partially inhibiting the production of PGI2 without inhibiting thromboxane generation systemically are unknown. These findings raised concern about the potential for Vioxx to predispose to thrombotic cardiovascular … events. Although there was not any evidence of an increased incidence of such events in the unblinded clinical trials of Vioxx to date, seven studies of Vioxx are currently ongoing. These studies are still blinded to treatment allocation, and comparisons of CV events in Vioxx treated patients compared to controls cannot be performed.
For comparison in his analysis, Dr Watson used placebo data from corresponding trials of other drugs, Fosamax and Proscar. The purpose of the analysis was stated as follows:
The purpose of this analysis was to provide information on which to base a recommendation to the Vioxx project team regarding the need for more formal monitoring of the risk of thrombotic CV serious adverse events … with Vioxx.
The “specific objective” was stated as follows:
The specific objective of the analysis was to evaluate the incidence of thrombotic [cardiovascular serious adverse events] among patients in the Vioxx Phase IIb and III [osteoarthritis] trials and their extensions, stratified by age and time period at risk. For purposes of comparison, the incidence of the same events was estimated with placebo control patients from Fosamax and Proscar clinical trials, and to the incidence of [cardiovascular disease] in a population‑based epidemiological study.
Dr Watson noted that the study had limitations, such as the estimation required in relation to data from ongoing studies, misclassification of events, and the potential for bias inherent in the use of historical controls. Nonetheless, Dr Watson concluded his report in the following terms:
Based on this analysis, the [cardiovascular serious adverse event] incidence rates in trials of Vioxx appear roughly consistent with what would be expected in the general population, and there is no clear evidence of consistently elevated risk compared to age‑adjusted placebo controls from Proscar and Fosamax trials. As a result, no change in the conduct of the Vioxx trials appears warranted based on present results. An analysis of [cardiovascular serious adverse] event rates in patients treated with Vioxx compared to those treated with Vioxx [sic] placebo/comparators is recommended when the trial databases are unblinded.
46 The results of Protocol 023 were published as Catella‑Lawson (1999). Dr Garret FitzGerald was one of the authors. Although not published until May 1999, I note, from a footnote to the article to which I next turn, that the authors of this (ie the Catella‑Lawson) article appear to have reported on the results of Protocol 023 at a vascular biology symposium in San Francisco in April 1998. The authors noted that the metabolite TX‑M was reduced in the NSAID arm, but not in either the MK‑966 arm or the placebo arm. This result was consistent with the sparing of COX-1 (and therefore of TXA2) by MK‑966, and was expected. But they also noted that the metabolite PGI‑M was reduced in both the MK‑966 and in the NSAID arms. This result was not expected. The authors said:
An additional finding of this study was the suggestion that extrarenal biosynthesis of prostacyclin also was mediated by COX-2. This was unexpected because COX-1, but not COX-2, is expressed constitutively by endothelial and vascular smooth muscle cells in vitro …
The authors suggested several explanations as to how it might be that COX-2 mediated the production of vascular prostacyclin, including the up‑regulation of the enzyme by laminar shear stress in the endothelium, the biosynthesis of prostacyclin via COX-2 in subjects of advanced age experiencing platelet activation and inflammation, and the up‑regulation of the enzyme in response to the shedding by activated platelets of microparticles containing arachidonic acid. However, it was the realisation that COX-2 may be responsible for the production of endothelial prostacyclin that effectively initiated the scientific debate that lies at the heart of this proceeding. The authors of this article raised the question whether the selective inhibition of COX-2, by turning off the production of prostacyclin in circumstances where it might otherwise be induced while leaving the production of thromboxane unaffected, might have a prothrombotic tendency. They said:
Presently, the implications of prostacyclin suppression in vivo are unclear. … Prostacyclin formation by the vasculature is of functional importance in limiting the response to a thrombotic insult in mice, and we have shown previously that urinary excretion of PGI‑M is increased in syndromes of platelet activation. … It remains to be established whether treatment with specific COX-2 inhibitors will suppress this response.
The hypothesis that, if the production of endothelial prostacyclin is blocked but that of platelet thromboxane is unaffected, an imbalance will result with possible thrombotic outcomes came to be known as the “FitzGerald hypothesis”.
47 Dr FitzGerald was also a co‑author of what has come to be regarded as a companion study to that published as Catella‑Lawson (1999). This study was conducted by the same group of researchers as were involved in the Catella‑Lawson study. It was published (a few months, it seems, before the Catella‑Lawson study) as McAdam (1999). The study was concerned to examine the effects of celecoxib on indices of COX-l dependent thromboxane and on systemic prostacyclin. It was found that both celecoxib and the NSAID comparator (ibuprofen) inhibited the production of prostacyclin, but that only the NSAID inhibited the production of thromboxane. The authors concluded:
Celecoxib exhibits relative rather than absolute biochemical selectivity for COX-2 ex vivo at doses tolerated in humans. Nonetheless, it does not modify TXA2‑dependent platelet aggregation, reflective of its modest inhibitory effect on COX-1. Ibuprofen and 800 mg celecoxib inhibit COX‑2 ex vivo and suppress urinary PGI‑M to a comparable degree. This appears to extend to the class of COX-2 inhibitors. It remains to be determined whether this effect is sustained during chronic dosing in age groups at risk for cardiovascular disease. If this is so, trials much larger than those necessary to detect efficacy and safety in arthritis … will be necessary to determine whether cardiovascular consequences of inhibiting PGI2 biosynthesis will modulate the anti‑inflammatory benefit to be derived from chronic administration of COX-2 inhibitors in humans.
Thus, this McAdam study advanced essentially the same hypothesis about cardiovascular risk as was then shortly to be advanced by Catella‑Lawson and colleagues.
The Board of Scientific Advisers’ Meeting
48 MRL referred consideration of the ramifications of Protocol 023 to its Board of Scientific Advisers, a panel of experts external to Merck who met once a year to review data from programs currently being undertaken. Members of the Board came from academic centres around the world. Two of them were Dr John Oates, an expert in prostaglandin biology from Vanderbilt University and Dr Myron Weisfeldt, a cardiologist from Columbia Presbyterian Medical Centre. Relevantly to present issues, the Board met over the period 3‑6 May 1998. The Board received a presentation from Dr Alan Nies, the head of the Vioxx project team within Merck (MK‑966 having, apparently, by then assumed its intended retail name). Because it may be taken to represent the thinking of MRL at the time about Vioxx, it is useful to refer to that presentation in some detail.
49 In his “Introduction”, Dr Nies said:
It is currently understood that prostaglandin synthesis in humans is catalyzed by at least two forms of cyclooxygenase, …COX-1…and…COX-2,…. COX-1 is constitutively expressed and enzymatically active in a variety of tissues including the stomach, intestine, kidneys and platelets. COX-1 may therefore be the isoform responsible for the physiological functions of prostanoids including gastric mucosal protection and normal platelet function.
In contrast to COX-1, COX-2 is not constitutively expressed in most tissues but rather is induced by mediators such as serum growth factors, cytokines, and mitogens. Synaptic activity has been shown to elevate COX-2 expression in rats. COX-2 has been detected in leukocytes, vascular smooth muscle cells, human rheumatoid synoviocytes, human amnion, and the brain. COX-2 is also expressed in the normal kidney, where it may be responsive to dietary salt intake. Colonic adenocarcinoma has also recently been associated with increased COX-2 expression. In inflammatory exudates and in tissue culture, COX-2 induction is inhibited by corticosteroids. Given the localization and pattern of induction, COX-2 is hypothesized to be primarily responsible for the synthesis of prostanoids that mediate responses to pathological processes such as the propagation of inflammation, pain, and fever.
Dr Nies referred to the gastrointestinal toxicity of traditional NSAIDs, and continued:
A specific COX-2 inhibitor would therefore have the potential to suppress the pathological effects of prostanoids (e.g., inflammation) without toxicity allegedly associated with inhibition of COX-1. If COX-1 derived prostanoids do not significantly contribute to the symptoms or pathogenesis of acute and chronic inflammation, then a selective COX‑2 inhibitor, free of dose limiting COX-l associated toxicity, may demonstrate comparable or possibly greater clinical efficacy than existing NSAIDs for a variety of clinical disorders.
50 Dr Nies explained, by reference to studies which had been performed, that Vioxx was a highly selective COX-2 inhibitor at the proposed therapeutic doses. He discussed the efficacy of Vioxx in relation to osteoarthritis, rheumatoid arthritis and fever, and as an analgesic. He then dealt with questions of safety, commencing with gastrointestinal safety and dealing with several other areas where Vioxx might be more or less safe than other drugs.
51 Finally, under the heading “Prostacyclin Metabolism”, Dr Nies said:
In the renal study described above in subjects 60 – 80 years old balanced on a 200 mmol sodium diet, the urinary excretion of several prostaglandin metabolites was measured. Indomethacin but not VIOXXTM reduced the excretion of 11‑dehydro thromboxane B2. This finding was expected since this enzymatic metabolite of thromboxane A2 largely reflects platelet COX-l activity in normal subjects and therefore should not be affected by COX-2 inhibition. On the other hand, the excretion of two metabolites of prostacyclin, 6‑keto PGF1α and 2,3, dinor‑6‑keto PGF1α (PG‑M) were reduced following treatment with indomethacin 50mg. t.i.d. or VIOXXTM 50 mg. QD for 14 days. Standard thinking is that PGIM is index of total body synthesis of prostacyclin and that 6‑keto PGF1α arises largely in the kidney (probably renal vasculature) but that some may arise from systemic sources. The reduction of 6‑keto PGFl1α by VIOXX™ was not unexpected since COX-2 is known to be in the kidney. However, the finding of an effect of COX-2 inhibition on the excretion of PGlM was entirely unexpected. Current knowledge would suggest that COX-2 is not an important enzyme in the synthesis of prostacyclin by normal endothelium. Therefore additional experimentation is required to understand these findings. Experiments are being performed by Merck Frosst and Safety Assessment to determine whether 1) the effect on PGIM is shared by other COX-2 inhibitors, 2) whether VIOXXTM alters the synthesis of PGI2 from PGH2, 3) whether VIOXX™ alters the metabolism of PGI2 to PGIM, and 4) whether the kidney is a potential source for all of the metabolites. The effect of lower doses of VIOXX™ (12.5 mg and 25 mg daily) on the excretion of PGIM in volunteers will be determined by Clinical Pharmacology. Also, acetaminophen has been reported to reduce the urinary excretion of PGIM without affecting thromboxane metabolite excretion and this finding will be confined by Clinical Pharmacology.
To assess the potential implication of these biochemical findings on cardiovascular health, the clinical team and epidemiology have analyzed the blinded VIOXX™ database for serious cardiovascular events. The analysis does not suggest a concern. An initial look at the unblinded safety data from the Phase III comparison study vs. diclofenac (approximately 1300 patients followed for 6 months) also does not indicate an excess of cardiovascular events in patients treated with VIOXX™. Additional analyses will be performed on unblinded data when available.
The renal study referred to was Protocol 023.
52 At the conclusion of their meeting, the Board prepared a memorandum recording their consideration of, and their recommendations in relation to, Merck’s Vioxx program. In the opening “Background” section of their memorandum, the Board noted as follows:
The Board notes that the freedom from gastrointestinal toxicity that provides such an advantage to Vioxx could lead to the use of doses that inhibit COX-2 to a greater degree than seen with current NSAlDs, and that this could lead to adverse effects of inhibiting COX-2 that are not predictable based [on] experience with current NSAIDs.
Under the heading “Cardiovascular Pathophysiology”, the Board said:
The Board addressed the potential for either benefits or adverse consequences of selective inhibition of COX-2 on coronary heart disease. The possible effects of COX-2 inhibition on three separate components of the process leading to coronary ischemic events were considered. These are:
1. The development of lipid‑rich coronary plaques
2. The destabilization of the cap of these plaques by inflammatory cells, making them “rupture prone”, and
3. The thromobotic occlusion of the vessel at the site of plaque rupture with ensuing consequences of ischemia.
Dealing with the first item, the Board noted that the formation of foam cells was central to the process of atherogenesis, and continued:
There is a growing body of evidence indicating that inflammatory disease is a risk factor for myocardial infarction, and such inflammatory processes almost certainly are accompanied by the release of cytokines that affect cells in the monocytic series in general, and COX-2 expression in particular. Accordingly, it is appropriate to ask whether COX-2 expression in monocyte‑macrophage‑foam cell development regulates the progression of the atherosclerosis that accompanies dyslipidemias, either positively or negatively. If this were the case, then inhibition of COX-2 might be expected to alter the process of plaque formation.
As to the second item, the Board said:
Recent evidence indicates that inflammatory cells are present in the region of the thinned‑out cap that covers atheromatous plaques, and it is thought that these inflammatory cells contribute to thinning the plaque at its margin that is the frequent site of plaque rupture. As the critical cells in this process are inflammatory cells, the possibility exists that the products of COX-2 could regulate thinning the cap of plaques and render them rupture‑prone.
53 It is the way the Board approached the third item that is of primary (although not singular) importance in the present case. It said:
After the rupture of a coronary plaque, factors which regulate thrombus formation and which regulate the occurrence of ischemic ventricular fibrillation are critical to the clinical outcome. The initial thrombus at the site of plaque‑rupture is platelet‑rich and is labile. Accordingly, it is susceptible to any anti‑aggregatory influences, as is illustrated by the effectiveness of GPIIb‑IIIa antagonist. Prostacyclin is the most potent endogenous inhibitor of platelet aggregation, and studies in animal models indicate that its participation in inhibiting platelet‑activation is substantial. This is further supported by the thrombotic events occurring in the IP receptor knockout mouse. Not only does prostacyclin inhibit platelet activation, but it also potently inhibits the development of ischemic ventricular fibrillation in the canine model of myocardial infarction. With in situ hybridization, the vessel wall is the predominant site of prostacylin‑synthase, which is also abundant in the uterus. Cell culture experiments demonstrate a high degree of prostacyclin biosynthesis capacity in both endothelial cells and in the vessel wall as a whole.
All of this indicates that the vessel wall is a primary site of prostacyclin biosynthesis in the body, a localization that coincides with function. Is the prostaglandin endoperoxide substrate for the prostacyclin synthase provided by COX-l, COX-2 or both? The answer to this question, as it relates to normal human vasculature and the vasculature in the foam cell environment is not clear: a highly specific antibody for COX‑2 has not yet been employed in the appropriate studies in human vessels. In the rat, following vascular injury, the increase in prostanoid biosynthesis is mediated in large part by increases in COX-2. COX-2 is also unregulated in endothelial cell cultures by shear stress, several cytokines, and hypoxia.
On this background of information regarding the anti‑platelet and anti‑fibrillatory effects of prostacyclin and its vascular localization, it has been found that Vioxx reduces the urinary excretion of the prostacyclin metabolite, 2,3‑dinor‑6-keto‑PGF.α. This is important data but it is not a basis for any conclusion. Rather, it should be taken as a basis for hypotheses that should be actively pursued.
One hypothesis would be that the excretion of the prostacyclin metabolite, 2,3‑dinor‑6‑keto‑PGF.α. does not reflect systemic/vascular prostacyclin biosynthesis. An alternative hypothesis is that prostacyclin biosynthesis in the vasculature is inhibited by Vioxx (without blocking production of thromboxane A2 by the platelet). By removing this potent inhibitor of platelet aggregation, the probability that a coronary plaque rupture would lead to myocardial infarction or ischemic ventricular fibrillation is enhanced.
More information is clearly needed to address these hypotheses and the other questions related to the possible influences of a COX-2 inhibitor on coronary morbidity and mortality.
54 The Board then proceeded to make its recommendations on the subject. It opined that it was unlikely that any of the individual trials with Vioxx would have sufficient power to determine whether coronary events were increased or decreased by COX-2 inhibition. It proposed, therefore, that coronary events be predetermined endpoints in all future controlled trials with Vioxx and other COX-2 inhibitors, and that those endpoints be assessed by a uniform set of criteria, to enable the performance of a “meta‑analysis” of events. Two of the trials then being conducted involved patients with rheumatoid arthritis and Alzheimer’s symptoms, and the Board suggested that there may already be a sufficient number of events to permit a meta‑analysis. The Board considered the utility of various metabolites of prostacyclin to indicate source, and therefore to answer the question whether the reduction observed in Protocol 023 was of prostacyclin in the kidney or prostacyclin in the vasculature. The Board touched also upon the possibility of certain animal studies.
55 Under the heading “The effect of COX-2 inhibitors in pathologic states of platelet activation in humans”, the Board said:
Sustained in vivo activation of platelets and a resulting increase in systemic biosynthesis of thromboxane A2 occur in a number of conditions associated with vessel wall disease in humans, including hypercholesterolemia, cigarette smoking, scleroderma, and ischemic limb disease. Prostacyclin biosynthesis is also increased in these conditions, suggesting that COX-2 may be induced. Assessment of the effect of COX-2 inhibition on in vivo platelet activation in one of these vessel wall diseases would provide valuable information on the consequence of loss of prostacyclin in a condition of increased platelet activation. If this does occur, MRL should be the first to know, and thereby be able to take the lead in developing the obvious strategy of the combination of very low‑dose aspirin (30‑40 mg daily) with a COX-2 inhibitor.
The Board’s memorandum concluded its treatment of the relevant subject in the following terms:
The above considerations regarding hypothetical adverse effects and potential unexpected therapeutic benefits of Vioxx are part of the scientific intelligence gathering appropriate to the development of any truly novel pharmacological entity, and should be addressed in parallel with the conclusion of the process of acquisition and analysis of the data that will place this drug in the hands of patients. The gain in safety achieved by the elimination of serious and fatal gastrointestinal toxicity will free patients from one of the most serious adverse effects in current drug therapy. Thus, there is a strong mandate for introduction of Vioxx into medical practice as soon as is feasible.
The CV SOP
56 There was a Vioxx Project Team meeting at MRL on 12 May 1998. Dr Nies reported that the meeting of scientific advisers had, amongst other things, recommended that MRL systematically collect data on cardiovascular events in all clinical trials of Vioxx. In what appears to be Dr Nies’ own commentary, the minutes of the meeting continued as follows:
To accomplish this task, an adjudication committee should be established and follow a formal plan. Alan Nies has asked Doug Watson to head this adjudication committee for [cardiovascular] events. The committee’s guidelines for operation would be similar to the adjudication procedures for [perforations, ulcerations and bleeds]. Discussions are in progress regarding the draft protocol for this analysis. The plan should begin immediately; perhaps, with the Alzheimer’s trial. Background: The consultants were in two ideological camps on this subject: 1) Since atherosclerosis is an inflammatory disease, patients should benefit from inhibition of COX-2 activity; 2) Based upon data on [prostacyclin] metabolism obtained for [Vioxx], it is conceivable that [Vioxx] could disturb the [endothelium‑platelet] interaction to favor platelet aggregation.
57 Consistently with the proceedings of the project team meeting on 12 May, on 15 May Dr Watson convened the first meeting of what was described as a task force to formulate a plan such as was proposed. The section of the minutes of that meeting headed “Introduction, Purpose and Goal” recorded Dr Watson as having said:
In MK‑0966 Protocol 023 (A Double‑Blind, Placebo‑Controlled, Parallel‑Group Study to Evaluate and Compare the Effects of L‑748,731 [MK‑0966], Indomethacin, and Placebo on Urinary Sodium Excretion and Other Parameters of Renal Function in Subjects Consuming a 200 mEq Sodium Diet), VIOXX (50 mg daily), and indomethacin (50 mg 3 times daily) were both found to significantly reduce the urinary excretion of PGI‑M compared to placebo. However, VIOXX had no effect on systemic thromboxane production (largely platelet deririved) as assessed by measurements of serum TxB2 and the urinary, 11‑dehydro‑thromboxane B2. It was felt that the reduction in urinary PGI‑M likely reflects both a reduction in renal PGI2 generation, as supported by an observed decrease in urinary 6‑keto PFG1α, and to some undefinable extent a decrease in systemic PGI2 synthesis. PGI2 is synthesized ubiquitously blood vessel walls from arachidonate derived from either the vessel wall or the platelet. It is the most potent of all inhibitors of platelet aggregation, and has vascular dilatory properties as well. Conversely, TXA2, a potent platelet aggregator and vasoconstrictor, is synthesized primarily by the platelet. The clinical implications of partially inhibiting the production of PGI2 without inhibiting thromboxane generation systemically are unknown. These findings raised concern about the potential for VIOXX to predispose to thrombotic cardiovascular (CV) events.
58 The result of the work of Dr Watson’s task force was the production of a formal plan called the Standard Operating Procedure for the Surveillance, Monitoring, and Adjudication of Acute Thromboembolic Vascular Events in Clinical Trials of COX-2 Specific Inhibitors. By way of shorthand, it was referred to as the “CV SOP”. It does not appear from the evidence when the first version of that plan was completed. The earliest version in the evidence is dated 18 February 1999, but that has been stamped “superseded”. A version dated 30 August 1999 is also in evidence, and it is that to which I shall refer. The executive summary included the following:
[The CV SOP] contains standard procedures to be used in all eligible trials for event surveillance, monitoring, documentation, and adjudication by a blinded, external, Vascular Event Committee (VEC). The purpose of this SOP is to provide standardized data for pooled analyses of the incidence of vascular events in patients treated with a [COX-2 specific inhibitor] compound and of patients treated with one or more comparator agents. These procedures apply to all trials of Vioxx … and future [COX-2 specific inhibitor] compounds which are of ≥4 weeks duration, and include all events occurring on treatment or within 14 days following the discontinuation of treatment. These include trials and trial extensions conducted by MRL … Trials which are done in normal, healthy volunteers, regardless of duration of the study, are excluded. This SOP is not restricted to specific indications.
The Vascular Event Committee is composed of three separate subspecialty committees: one each for cardiac events, cerebrovascular events, and peripheral (pulmonary, abdominal/pelvic and extremities) vascular events. The members of the three committees are, respectively, cardiologists, neurologists, and vascular specialists who are expert in the treatment of ischemic syndromes as well as the medical aspects of clinical trials. Each subspecialty committee contains three active members. Adjudications are performed in a blinded manner using prespecified event definitions and adjudication guidelines.
The CV SOP provided that vascular events of a thrombotic or embolic nature occurring in the arterial vascular bed were of primary interest. They included fatal and non‑fatal acute myocardial infarction, unstable angina pectoris, fatal and non‑fatal ischaemic stroke, other fatal and non‑fatal acute arterial thromboembolism, sudden cardiac death, and resuscitated cardiac arrest. Vascular events of secondary interest included fatal and non‑fatal pulmonary embolism, fatal and non‑fatal venous thrombosis, non‑fatal cardiac (atrial or ventricular) thrombosis and transient ischaemic attack. Henceforth in these reasons, in the context of studies and trials conducted by Merck under the CV SOP, I shall refer to all these events collectively as “CVT events”.
59 It will be recalled that the Board of Scientific Advisers had been of the view that it was unlikely that any individual trial of Vioxx would (at least with respect to cardiovascular concerns) have sufficient power to yield useful results. Consistently with that, the CV SOP was a procedure for the analysis of pooled results of multiple trials and studies. It provided:
Analysis will be performed on blocks of studies grouped according to study design, indication, and the timing of their completion. The objective of the statistical analysis is to estimate the incidence of acute Thromboembolic CV events in patients treated with a specific C‑2SI compound and that of patients treated with one or more comparator agents. No formal comparisons among treatment group rates will be performed. Within a given analysis, for purposes of computing incidence rates, only the first event in each subject will be counted. The incidence rate for each group will be expressed as a fraction whose numerator is the number of subjects in a group experiencing an event, and the denominator is the total number of person years at risk for all subjects in the group.
60 There are three aspects of the CV SOP to which attention should be drawn at this stage. The first is that it provided for events of interest to be counted if they occurred while a patient was on treatment, or within 14 days of discontinuation. In every trial of a drug, there will be a predetermined period over which patients in the trial are expected to take the drug (or comparator). It may be, for example, for the period of the trial as such. Or it may be for a set period, even if that is a shorter period than that of the trial. A trial may, for example, be planned to run until a certain number of events of interest have occurred: in such a case, it will not be known beforehand how long the trial will take. However things are done, it is said that the “intention‑to‑treat” (“ITT”) period is the period over which it is (prospectively) intended that each patient will take his or her therapy. However, it is an unhappy fact that many patients discontinue taking their therapy, whether by way of formal withdrawal from the trial or otherwise. Researchers have the problem of deciding whether to “count” an event of interest if it occurs only when the patient concerned is actually taking his or her therapy (known as the “per‑protocol” or “on‑drug” approach) or if it occurs at any time within the ITT period. In the case of the CV SOP, the approach taken, as I say, was to count events which occurred up to the 14th day after discontinuation. Although this approach was controversial in the case, counsel for the applicant did not submit that the CV SOP itself was deficient in this respect.
61 The second aspect of the CV SOP presently requiring mention is that the essence of its genesis was the view of the Board of Scientific Advisers that individual trials of Vioxx would, because of smallness of numbers, be unlikely to have sufficient statistical power to yield useful results on cardiovascular events. The point of the CV SOP was, therefore, to conduct a meta‑analysis of the cardiovascular events recorded in many trials and studies. It was, accordingly, proposed that formal comparisons between the arms of individual trials not be carried out. As will become apparent, this was a guideline occasionally honoured in the breach.
62 The third aspect relates to the process of adjudication for which the CV SOP provided. I have already referred to the role of investigators in clinical trials. The classification of adverse events according to predetermined clinical categories was, however, to be the task of specialist, blinded, committees. The extract set out at para 58 above refers to them. However, in the case of some of the trials which had already been completed, or substantially completed, before the promulgation of the CV SOP, there had not been a cardiovascular adjudication process in place at the time. Although the CV SOP applied only to trials which began in the second quarter of 1998 or later, in some of its reports to the FDA Merck relied also on the results of earlier trials, and in such instances, of necessity, investigator‑reported events were used.
The US New Drug Application for Vioxx
63 Merck lodged its New Drug Application for Vioxx with the FDA on 23 November 1998. Although the correspondence covering the application is in evidence, the application itself (the paper copy of which ran to some 278 volumes) is not. The correspondence contained a compendious statement of the expected characteristics of Vioxx, and of its clinical advantages over traditional NSAIDs:
Vioxx is a potent, orally active cyclooxygenase‑2 (COX-2) specific inhibitor. This COX-2 specific inhibition is demonstrable within, and significantly above, the clinical dose range. Cyclooxygenase is responsible for the generation of prostaglandins, which are potent biological mediators involved in diverse physiological functions as well as pathologic conditions. Two isoforms of cyclooxygenase have been identified: cyclooxygenase‑1 (COX-1) cyclooxygenase‑2 (COX-2). COX-1 is constitutively expressed and enzymatically active in various tissues, including the stomach, intestines, kidneys and platelets. Evidence suggests that COX-1 is responsible for prostaglandin‑mediated physiologic functions such as gastric cytoprotection and platelet aggregation. Inhibition of COX-1 in the gastric mucosa by nonspecific cyclooxygenase inhibitors (commonly known as NSAIDS) has been associated with gastric damage. In contrast, COX-2 is constitutively expressed in only a limited number of tissues, including the brain and kidney, and is the inducible isoform of the enzyme that has been shown to be up regulated by proinflammatory stimuli. Based on patterns of expression and localization, COX-2 has been postulated to be primarily responsible for the synthesis of prostanoid mediators of pain, inflammation, and fever. The efficacy of VIOXXTM is due to its specific inhibition of COX-2. Because VIOXXTM does not inhibit COX-1 within and significantly above the clinical dose range, it has a safety profile in the GI tract superior to NSAIDS and similar to placebo, when administered in the clinical dose range.
64 Dr Reicin said that, in the United States, a New Drug Application includes the data from all animal and human studies conducted in relation to the proposed new drug, and analyses of those data, as well as information about how the drug works in the body and how it is manufactured. In the case of Vioxx, the New Drug Application and the safety update report contained data from 60 Phase I, II, and III clinical trials involving approximately 10,000 individuals. It included summaries as to efficacy and safety data, an integrated assessment of the benefits versus risks relationship, proposed labelling, case report tabulations, and individual case report forms for each patient who died during a clinical study or who did not complete the study because of an adverse event. At the time, there were industry standards as to the extent and duration of treatment needed to provide a safety data base for drugs intended for long‑term treatment. Those standards recommended that approximately 1500 individuals should be treated with a new drug like Vioxx. Dr Reicin said that a cumulative total of 6,062 individuals received Vioxx in clinical trials conducted prior to approval. The standards recommended that between 300 and 600 patients be exposed for more than six months. Dr Reicin said that a cumulative total of 1,471 patients received Vioxx for at least that long. The standards recommended that 100 patients be exposed for a minimum of one year. Dr Reicin said that a cumulative total of 824 patients received Vioxx for one year or longer, and that 94 patients had taken Vioxx for two years or longer prior to its approval. She added that the studies referred to in the New Drug Application compared Vioxx to the traditional NSAIDs ibuprofen, diclofenac, and nabumetone. There were also some placebo‑controlled studies of Vioxx, but Dr Reicin explained that these studies were limited in duration because it was not possible to keep patients with chronic painful arthritis on a placebo for very long.
65 The New Drug Application contained a section – called the Integrated Summary of Safety – which combined the adverse experiences from all of the clinical trials referred to. This included an analysis of adverse experiences recorded in every body system, including the cardiovascular system (and adverse experiences of any kind that occurred while a person was taking the drug, whether or not considered to be drug‑related). In the analysis of thromboembolic cardiovascular events, it was reported that the incidence of serious cardiovascular adverse experiences was not statistically different as between treatment groups. It was also reported that groups taking Vioxx and those taking placebo and NSAID comparators had similar rates of thromboembolic cardiovascular adverse experiences generally, regardless of their seriousness.
66 Dr Reicin said that the FDA would appoint an advisory committee of external experts in a variety of disciplines to assist it in deciding on certain drug applications. She said that, on 20 April 1999, the FDA’s Arthritis Advisory Committee met to discuss the Vioxx application. Representatives from Merck and the FDA gave presentations, after which the committee held an open public hearing and a discussion and questions session. The committee recommended unanimously that Vioxx 12.5 mg and 25 mg be approved for the relief of the signs and symptoms of osteoarthritis, the management of acute pain, and the treatment of primary dysmenorrhea.
67 The FDA approved Vioxx by letter to Merck dated 20 May 1999. In that letter, the FDA said:
We have completed the review of this application, as amended, and have concluded that adequate information has been presented to demonstrate that the drug product is safe and effective for use as recommended in the enclosed labeling text. Accordingly, the application is approved effective on the date of this letter.
The final printed labeling (FPL) must be identical to the enclosed labeling (text for the package insert). Marketing the product with FPL that is not identical to the approved labeling text may render the product misbranded and an unapproved new drug….
If additional information relating to the safety or effectiveness of this drug becomes available, revision of the labeling may be required….
Please note that any advertising and/or promotional activity of this product will be considered false and/or misleading … if it presents suggestions or representations that COX-2 selectivity confers on the product any claims of safety beyond what has been demonstrated in clinical studies and presented in the approved labeling. Additionally, promotional activities that make or imply comparative claims about the frequency of clinically serious gastrointestinal … events compared to groups of NSAIDs or specific NSAIDs will be considered false and/or misleading without differences having been demonstrated in adequate, well‑controlled studies.
Dr Oates’ correspondence
68 On 13 August 1999, Dr Oates wrote to the Head of MRL, Dr Ed Scolnick, (with a copy to Dr Nies) to acquaint Merck with information which he had received “regarding thrombotic complications occurring in patients with the antiphospholipid syndrome very soon after initiating therapy with celecoxib”. Dr Oates referred to three patients. The first was a woman in her 40s with a connective tissue disorder. She had a positive rheumatoid factor and Raynaud’s phenomenon. She had features of Sjogren’s syndrome and systemic lupus erythematosus. She also had atherosclerosis on angiography. After the second dose of celecoxib, the patient developed pain in the foot, which was diagnosed as a clot in a distal artery.
69 The second patient was a woman of about 30 years with a diagnosis of systemic lupus erythematosus. It was known that she had an elevated IgG anticardiolipin antibody. After the third dose of celecoxib, all five toes on the left foot abruptly became cyanotic in a fashion typical of microvascular ischaemia.
70 The third patient was a woman of 56 years who had been diagnosed with scleroderma in 1995, associated with pulmonary hypertension and a positive lupus anticoagulant test. Dr Oates continued, with reference to this third patient:
She developed an ulnar artery thrombosis in May of 1997 and was placed on warfarin. Her PT‑INR was maintained in the 2.0‑2.5 range. In April of 1999, she developed leg pain and was placed on a selective COX-2 inhibitor. After 2‑4 doses, she developed dyspnea. She reported to the emergency room two days later. A V/Q scan was interpreted as “high probability for pulmonary embolism.” The prothrombin time INR was 2.5 on admission. Cardiac and lower extremity ultrasounds failed to reveal a source. She was treated with heparin, and a follow‑up V IQ scan was read as “low probability for pulmonary embolism.”
After the patient had stabilized and after the heparin had been discontinued, four separate urines were collected for analysis of the prostacyclin metabolite, 2,3‑dinor 6‑ketoF1α and 11‑dehydrothromboxane B2 (a urinary metabolite of thromboxane A2). The levels of 11‑dehydrothromboxane B2 were, as would be expected in this disorder, more than 6‑fold above the upper limit of normal. Importantly, the levels of the prostacyclin metabolite also were increased (by 48‑74%), hypothetically reflecting induction of COX-2 in the vessel wall in response to the underlying thrombotic state. …
The findings in this patient are consistent with those previously reported for patients with the antiphospholipid syndrome … The findings of an increased excretion of prostacyclin metabolite in these patients is in line with such an increase seen in other disorders of excessive platelet activation (e.g. cigarette smoking).
71 Dr Oates noted that these patients suffered from a disease in which thrombosis was “a characteristic complication”, adding that their problems “may be only coincidentally related to the COX-2 inhibitor”. He continued:
However, the occurrence of the thrombotic complications so quickly after initiation of the drug raises the probability of causality. Clearly no conclusions may be made at this time.
The purpose of this communication is a “heads up” so that Merck will be fully aware of these developments and can have an opportunity to make any plans that are relevant to them.
Dr Oates then discussed the possible advantages to Merck if it were to develop a rofecoxib formulation that contained a low‑dose, slow‑release, aspirin component. He concluded his letter with this paragraph:
In summary, I am conveying information on three patients with the anti‑phospholipid syndrome who had onset of a thrombotic episode very soon after initiating treatment with celecoxib. Because thrombosis is a part of a natural history of the antiphospholipid syndrome, these occurrences may be purely coincidental. However, the temporal relationship as well as the biologic plausibility at least raise the question of whether there may be causality. I convey these to you with full knowledge of the fate that often befalls the messenger. However, I wish to assure that Merck is informed as well or better than Searle on this issue. If I could be helpful in any of your considerations, please let me know.
One of the cardiologists called by the respondents, Prof Douglas Vaughan, agreed that Dr Oates’ letter was predicated on the FitzGerald hypothesis being at least a plausible theory at the time.
The VIGOR trial
72 In 1998, MRL commenced the design of a major study into Vioxx, the Vioxx Gastrointestinal Outcomes Research trial, or “VIGOR”, as it was called. It was a prospective, double‑blind RCT of patients with rheumatoid arthritis. A “double‑blind” trial is one in which neither the patients taking the treatment nor the investigators administering the treatment and reporting on outcomes know which treatment is being taken in any particular case. Dr Reicin was the clinical monitor for the study. As such, she had primary responsibility for drafting the study design and protocol. Dr Reicin said that the trial was designed to examine whether Vioxx, by comparison with a traditional NSAID, significantly reduced the risk of gastrointestinal perforations, ulcers and bleeds.
73 Dr Reicin (and MRL) took the view that it would not be clinically appropriate to include a placebo arm in a lengthy rheumatoid arthritis study, since that would leave patients in that arm effectively untreated. Ultimately, naproxen was selected as the comparator because, first, MRL had already tested Vioxx extensively against ibuprofen, diclofenac, and nabumetone (and VIGOR presented an opportunity to collect data based on a comparison between Vioxx and another traditional NSAID), secondly, naproxen at a dose of 500 mg twice daily was the most commonly used NSAID treatment for rheumatoid arthritis in the United States, and thirdly, naproxen needed to be taken only twice a day, whereas ibuprofen and the form of diclofenac used in MRL’s Phase III studies needed to be taken three to four times a day (that is to say, the view was taken that naproxen would provide the patients in the study with a less complicated regimen, and make it more likely that they would be compliant).
74 In designing the VIGOR trial, MRL was faced with the same issues as to the taking of aspirin as I have referred to in paras 38‑41 above. It decided to exclude patients who required low‑dose aspirin from the trial altogether, because the trial was intended to test whether the use of a COX-2 selective inhibitor would provide enhanced gastrointestinal safety compared with a drug that inhibited both COX-l and COX-2. To allow patients in the Vioxx arm to take aspirin (a significant inhibitor of COX-1) during the trial would, quite evidently, frustrate the achievement of that purpose. This issue was one of a number discussed between representatives of MRL, including Dr Reicin, and representatives of the FDA at a meeting on 5 November 1998. The FDA agreed that the trial could proceed on the basis indicated, but made it clear that, if there were no other trial of, or acceptable evidence about, the concurrent consumption of aspirin and Vioxx, a suitable disclaimer would eventually have to appear on the label.
75 Also discussed with the FDA was the dose of Vioxx that should be used in the trial. Dr Reicin said that it was apparent from experience with traditional NSAIDs that some gastrointestinal side-effects were dose‑related. The FDA indicated that the dose of Vioxx to be used in the trial should be twice the anticipated dose in practice to allow for a margin of safety, and to allow for “dosage creep” sometimes observed in clinical practice (ie the tendency of some patients to take, or of some physicians to prescribe, higher than the recommended doses). The FDA also indicated that MRL needed to prove that the gastrointestinal benefit of Vioxx would be maintained even if the dose were double the maximum therapeutic dose for chronic conditions. The intended therapeutic dose was 25 mg/day, so the dose selected for the trial was 50 mg/day.
76 After screening, 8,076 rheumatoid arthritis patients were enrolled for the VIGOR trial. Of these, 4,047 were randomly assigned to receive Vioxx and 4,029 were so assigned to receive naproxen, from 301 clinical sites in 22 countries, including Australia. The VIGOR trial commenced on 6 January 1999. Over the course of the trial, the median length of time during which patients were taking their assigned medications was 9 months. Across both arms of the trial, about 80% of the patients were women, and the median age of patients was 58 years.
77 For the VIGOR trial, a Data Safety Monitoring Board was constituted, under the chairmanship of Dr Michael Weinblatt. This was a group of experts, external to Merck, whose role was to monitor safety data as they became available during the course of the trial. Data from the trial were to be provided to the board by reference to the two treatment groups, identified only as treatment A and treatment B. In every other respect, members of the board had access to all the data as they progressively emerged. The board was, in the lexicon of such trials, “unblinded” to the data. The purpose of that was to enable the board to fulfil its function of protecting patient safety during the course of the trial. Had the board, for example, observed a significant disparity in the occurrence of adverse events as between the two arms of the trial, it had the power to decide that the trial should be halted. Others working on the trial would remain “blinded” to the data until the trial was over. The purpose of that was to ensure that bias was not inadvertently introduced into the trial. There was only one member of the board who was not external to Merck, Dr Deborah Shapiro, a statistician who assisted the board by providing them with the required data and statistical analyses. Although “unblinded” like other members of the board, Dr Shapiro was under a duty of confidence not to disclose details of the emerging results of the trial to persons who were not on the board.
78 In December 1999, Dr Weinblatt recommended that Merck develop a plan to analyse the adjudicated cardiovascular events from VIGOR separately from the CV SOP. Dr Reicin initially resisted this suggestion upon the basis that such an analysis would be statistically underpowered, that is, there would not be enough events to permit any meaningful conclusions to be drawn. However, Dr Weinblatt persisted in his request, and MRL agreed to it in February 2000. As an addendum to the VIGOR Data Analysis Plan, MRL introduced a Plan for the Adjudication and Analysis of Serious Vascular Events. This plan specified what events would be included in the analysis, the cut‑off date for the inclusion of those events in the analysis, and how the data would be analysed. Events that were reported after the cut‑off date would be adjudicated, and the resulting information would be available, but would not be included in the analysis as such. The cut‑off date for gastrointestinal events was about a month later than that for vascular events, for the reason, according to Dr Reicin, that, in the case of the latter, it took longer to obtain the hospital and medical records that were needed to adjudicate those events.
The preliminary results of VIGOR
79 9 March 2000 was the date by which all patients in the VIGOR trial were to have completed their end‑of‑study visit. On that day, a small group of Merck personnel, including Dr Reicin, were unblinded to the preliminary results to facilitate the preparation of a summary for submission to regulatory agencies (principally the FDA). I shall refer to the actual results in some detail below. It is sufficient here to note that, as Merck had hoped, patients in the Vioxx arm of the trial had encountered significantly fewer serious gastrointestinal adverse events than had those in the naproxen arm. However, patients in the Vioxx arm had experienced significantly more serious thromboembolic cardiovascular adverse events than had those in the naproxen arm. When Dr Reicin saw these results, her first reaction was a concern that Vioxx was increasing the risk of heart attacks because it was prothrombotic. She agreed with counsel for the applicant that a great deal of work went into trying to understand the results.
80 Dr Scolnick had a similar reaction. On 9 March 2000, he sent an email to Dr Reicin, Dr Shapiro and Dr Nies, in which he noted that the cardiovascular events were “clearly there”. He observed that there was “no obvious correlate”, and continued:
It is important to find out about the cases that Oates told us about. When we present in May we should present those also if he will let us so it is clear to the world that this is class effect.
The “cases that Oates told us about” were the three patients to which I have referred in paras 68‑71 above. By the term “class effect”, Dr Scolnick meant that the effect observed in the VIGOR trial was not peculiar to Vioxx, but was common to coxibs as a class. In his email of 9 March, Dr Scolnick made a number of other brief observations about possible relationships that might be involved in the cardiovascular results of the VIGOR trial. In the course of that, Dr Scolnick said:
It is a shame but it is a low incidence and it is mechanism based as we worried it was. Oates and Alan and [B]arry were right about the metabolite meanings ie urine Pg data.
The respondents did not call Dr Scolnick, and I believe I am entitled to infer that the words “mechanism based” and “the metabolite meanings ie urine Pg data” were a reference to the FitzGerald hypothesis.
81 In a reply to Dr Scolnick on 10 March 2000, Dr Reicin attached the abstract of a paper presented at a meeting or conference of the American College of Rheumatology in November 1999. The abstract read in part as follows:
Prostaglandins and thromboxanes are regulators of platelet and endothelial cell function. Activated platelets synthesize thromboxane A2, which is a potent platelet aggregant and vasoconstrictor. Prostacyclin, which is primarily synthesized by endothelial cells, inhibits platelet activation by increasing cyclic AMP levels, and induces vasodilation. A multi‑enzyme pathway that includes the cyclooxygenase (COX) enzymes is responsible for synthesis of prostanoids. There are two COX isoforms, COX-l and COX-2. COX-l is constitutively expressed in most tissues and is the only isoform in platelets. The anti‑platelet effects of aspirin and other NSAIDs are mediated through inhibition of COX-1‑generated thromboxane production. Celecoxib is a specific inhibitor of COX-2 approved for treatment of osteoarthritis and rheumatoid arthritis. Unlike aspirin and NSAIDs, specific COX-2 inhibitors do not inhibit platelet activation and thromboxane production. However, similar to NSAIDS, these agents reduce systemic production of prostacyclin. The resulting imbalance between thromboxane and prostacyclin could result in an alteration of the coagulation system. Patients with systemic lupus erythematosus (SLE), who may be predisposed to thrombosis and vasculopathy, often have arthritis requiring treatment with anti‑inflammatory agents. We report two patients with active SLE, as determined by elevated anti‑ds‑DNA antibodies and depressed complement levels, and high titer anti‑cardiolipin antibodies that developed lower extremity ischemic events shortly after the initiation of treatment with celecoxib. Both patients also has [sic] reduced free protein S antigen that could contribute to their pro‑thrombotic state. These data suggest that patients with underlying risk factors for thrombosis may experience adverse thrombotic events following treatment with specific COX-2 inhibitors. These agents should be used with caution, or low‑dose aspirin used concomitantly, in patients with SLE and risk factors for thrombosis.
82 As far as they went, the VIGOR results only revealed a difference between Vioxx and naproxen. One of the first things to which Dr Reicin turned her mind was the possibility that the results might be explained by the cardioprotective effect of naproxen rather than by the prothrombotic effect of Vioxx. She recalled the 1993 article to which she had referred in the endorsement she made on Dr Musliner’s draft protocol in February 1997 (Brochier 1993: see para 40 above). She made arrangements to have a copy of that article forwarded to her urgently, and it arrived on the evening of Sunday 12 March 2000. The author described flurbiprofen as an “NSAID with potent antiplatelet properties”. In the study on which the article was based, the six‑month reinfarction rate was 3% in the flurbiprofen arm and 24% in the placebo arm. The difference was significant. Dr Reicin said that the results observed in the flurbiprofen arm in the Brochier Study “were within the range of reduction that we saw in the naproxen group in VIGOR”. At 1.20 am on 13 March 2000, Dr Reicin sent a copy of the abstract for the flurbiprofen article to Drs Scolnick and Nies, commenting that it was the only study she could find which assessed the potential cardioprotective effect of an NSAID.
83 Dr Reicin gave evidence that she did in fact find another study on that subject: Fornaro (1993). The abstract for the article stated that “the results of the study indicate that indobufen may reduce the risk of ischaemic events in patients with heart disease associated with an increased risk of embolism.”
84 On the afternoon of 13 March 2000, Dr Reicin sent Dr Shapiro a spreadsheet of the cardiovascular events observed in the VIGOR trial. The spreadsheet itself is not in evidence, but Dr Reicin’s covering memorandum is. She informed Dr Shapiro that events which had occurred more than 14 days after the last dose of therapy were excluded. Dr Shapiro replied:
Unlike the GI events where it seems legitimate to delete events >14 days after study discontinuation, it is traditional and considered valid to do true intention to treat in cardiovascular trials and we typically would not delete events that occurred >14 days after the last dose of study therapy. I’m concerned that these events are considered ineligible for adjudication.
Dr Reicin’s response, sent at 12.24 am on 14 March 2000, was as follows:
I knew you would say this – although that is how the … adjudication plan was set up. Remember that WAES includes everything. I don’t think this affects that many patients. You were not on today’s teleconference but by the end of next week so many people will be unblinded it is hard to imagine that the adjudication committee won’t be tainted as well making this whole exercise fraught with problems – did you look at the events adjudicated already? I assume that it changes nothing.
85 It appears that there was a meeting to discuss the actions that were necessary in light of the results of VIGOR on 15 March 2000. I so infer from the terms of an email sent on the afternoon of 14 March 2000 by David Blois (whom Dr Reicin described as a senior person in regulatory affairs at Merck) to addressees who included Drs Scolnick, Reicin, Nies and Shapiro. The email contained a “to do list”, which was said to be “a good starting point for our discussions tomorrow”. Items on the list included “list of studies with rofecoxib …”; “protocol modifications?? allow low dose aspirin? other?”; “complete writing summary report and submit to agencies and others?”; “label modifications for the CV events”; “How to handle Safety Update report?”; “how to handle CV adjudication committee? what/when to talk to them? what should be their future role?”; and “any message balanced with GI outcome data? effect on publication?”. Dr Reicin could not recall whether she attended any such meeting on 15 March.
86 In their attempts to understand the cardiovascular results of the VIGOR trial, the scientists at MRL reviewed much of the data that had by then been collected in the earlier trials and studies of Vioxx. At that time MRL also had two ongoing placebo‑controlled trials for Vioxx in elderly patients with Alzheimer’s disease and early onset dementia. MRL was collecting and adjudicating cardiovascular events from those studies as part of the CV SOP. On 12 March 2000, the scientists reviewing the VIGOR results were unblinded to the cardiovascular data only in those trials. Those data revealed no statistically significant difference in the incidence of thrombotic cardiovascular events as between the Vioxx arms and the placebo arms.
87 The scientists also reviewed the data from the Phase IIb/III osteoarthritis trials to see if something had been missed. They performed a pooled cardiovascular analysis on the osteoarthritis data set of over 5,000 patients. On 16 March 2000, Dr Shapiro sent Dr Reicin, Dr Eliav Barr (Director, Biologics Clinical Research at MRL) and Dr Len Oppenheimer (a biostatistician to whom Dr Shapiro reported) an email with the relevant data from the osteoarthritis trials. Dr Reicin forwarded this information to Drs Scolnick and Nies (and to others) on the evening of the same day. The information itself is not in evidence, but Dr Reicin’s memorandum is. She said:
Attached are the final numbers from the Phase III OA data. The numbers confirm my “back of the envelope calculations”. VIOXX and Diclofenac are identical. Ibuprofen has a lower rate of events but the exposure and number of events are small (consistent with its antiplatelet effects?). The next e‑mail I will send shows data compared to placebo. Vioxx and placebo are similar and ibuprofen is lower than both vioxx and placebo – but again the # of events and exposure if [sic] very very small.
In her oral evidence, Dr Reicin said that the similarity of the rates of serious cardiovascular events for patients taking Vioxx and for patients taking NSAID comparators was “an important piece of information” for her. Shown her memorandum of 16 March 2000 under cross‑examination, Dr Reicin recalled that her view at the time was that ibuprofen was “completely non‑selective in terms of COX-1 and COX-2 inhibition but diclofenac in some … doses could be a little bit selective …”
88 On 16 March 2000, Dr Barr proposed that the data from VIGOR might be examined by reference to characteristics of the subject patients which had not previously been of interest, but which, apparently, were recorded. In an email to Drs Shapiro and Reicin, Dr Barr proposed that the experiences of patients who were smokers, and who were taking hormone replacement therapy, might be examined. Dr Reicin replied to Dr Shapiro, with a copy to Dr Oppenheimer, in the following terms:
Both smoking and HRT seemed to exaggerate the imbalance. Kinda supports the hypothesis that patients with a “prothrombotic” state are either more sensitive to the cardioprotective effects of aspirin/naproxen or the negative effects of vioxx
In an email in reply, Dr Oppenheimer made the following contribution:
If we look at the data and combine the “strong” results and then calculate the relative risk and find it to be “even stronger” that’s not “proving the hypothesis”---I’m not saying we shouldn’t be doing those calculations---but that we should exercise some caution until we have other truly independent results and information to actually confirm the hypothesis.
On 17 March 2000, Dr Barr replied to Drs Shapiro, Reicin and Oppenheimer, stating that he understood the “point” made by the latter. He continued:
The idea here is hypothesis generation, not proof of concept. All of these are post‑hoc analyses, and therefore suspect. The adjudication (and preadjudication) CV SAE analysis demonstrates that there is a greater incidence of [cardiovascular serious adverse events] in patients taking rofecoxib vs. naproxen in the VIGOR study.
There are several major questions that we are trying to answer from the VIGOR database:
1. Are the VIGOR results real? That is, are there KNOWN cardiovascular risk factors that are imbalanced between treatment groups that can explain the imbalance?
The CV adjudication confirms that there is a real problem in VIGOR. The results are not a consequence of misclassification and potential biases that may have occurred due to non‑thrombotic issues related to rofecoxib, such as increased edema and hypertension causing an increased scrutiny of the cardiovascular status of the rofecoxib patients relative to the naproxen patients that led to more procedures and more “MIs” – this is what happened in FIT. The question of baseline imbalances is certainly more problematic statistically: if you look enough, you’ll find something, so you may be fooled. So we all have to take everything with a grain of salt.
2. If the results cannot be explained by a chance demographic imbalance, is there an easily identifiable segment of the RA population that is of particularly high risk? Conversely, is there a segment of the RA population that is not at risk? This has obvious public health implications.
[Dr Shapiro] has already done an analysis of CV risk factors vs. events, and found that there does not appear to be segregation of effect in the high risk patients; I believe the data because it is internally consistent: event rates go up with numbers of risk factors, and the incidences are consistent with the CV literature in higher risk patients.
So, I want to know whether a different set of related, known risks have the same effect. Given the prothrombotic hypothesis being circulated, it is of interest.
If we find something in the VIGOR dataset, it would obviously be helpful if we can confirm it in other datasets; however, these kinds of analyses may cause more problems than they solve.
89 As will be apparent from the above communications, the information available to Dr Reicin and the scientists at MRL in the 10 days or so following 9 March 2000 was neither consistent nor, in a number of respects, satisfactory. As Dr Reicin explained in her evidence, VIGOR showed a higher cardiovascular event rate for subjects on Vioxx than for subjects on naproxen. Yet, the Phase IIb/III osteoarthritis studies showed similar event rates for subjects on Vioxx and for subjects on the NSAIDs ibuprofen, diclofenac, and nabumetone. And the Alzheimer’s studies showed similar event rates for subjects on Vioxx and placebo. Dr Reicin explained her state of puzzlement at the time in her witness statement as follows:
If Vioxx were increasing the risk of heart attacks, I would have expected that the event rates would have been higher on Vioxx than placebo in the Alzheimer’s studies, but that was not the case. If NSAIDs were cardioprotective (as we had hypothesized even before I was first working on the GI outcome study in early 1997), I would have expected that the event rates would have been higher on Vioxx than the NSAIDs ibuprofen and diclofenac, but that was not the case.
This unsatisfactory state of affairs subsisted, I would infer, until at least the exchanges to which I have referred in the previous paragraph. Then, probably about 17 March 2000 (Dr Reicin was unable, despite being pressed, to put a date on it) Dr Reicin had what she described in her evidence as an “ah‑hah moment” which had, to her, at least the obvious potential to resolve this state of puzzlement.
The significance of Protocol 061
90 While working through the data in March 2000, Dr Reicin spoke to Dr Barry Gertz, a clinical pharmacologist and endocrinologist who was then head of clinical pharmacology at MRL, about the VIGOR results and the confusing data sets with which she was confronted. Dr Gertz referred to pharmacological data on naproxen that MRL had gathered during the Vioxx clinical development program, and told Dr Reicin that, in his view, the data sets actually made sense. As I have mentioned earlier in these reasons, the inhibitory effect of NSAIDs (other than aspirin) upon platelet COX-1 is reversible. That is to say, as the concentration of the drug declines over a period of time (eg by excretion), the extent of inhibition of the enzyme will also decline. In his meeting with Dr Reicin, Dr Gertz produced data from a 1998 study known as Protocol 061 that illustrated that naproxen differed from ibuprofen and diclofenac in its ability to sustain the inhibition of platelet COX-1 over a period of hours. In that study, bleeding times and platelet clumping had been tested in patients taking Vioxx (12.5 mg and 25 mg once daily), meloxicam (15 mg once daily), ibuprofen (800 mg 3 times daily), naproxen sodium (550 mg twice daily – the equivalent of 500 mg of naproxen), diclofenac (50 mg three times daily) or placebo. A blood sample was taken as a baseline measure before the patients started taking the medication or placebo to which they were randomised. The patients took their medication or placebo for a period of 5 days. On the morning of the sixth day, the patients had a blood sample taken, and then took a further, and final, dose of medication or placebo. Then blood samples were taken two, four and eight hours later. When analysing the data, the baseline measurement was compared to the measurements at 0 hours, 2 hours, 4 hours, and 8 hours on the sixth day.
91 A number of tests were conducted in Protocol 061, two of which were mentioned by Dr Reicin. The first involved an examination of the effect of the different treatments on bleeding time, as a measure of their ability to inhibit arachidonic acid‑induced platelet aggregation. The report of the study which Dr Gertz showed to Dr Reicin contained the following graph:
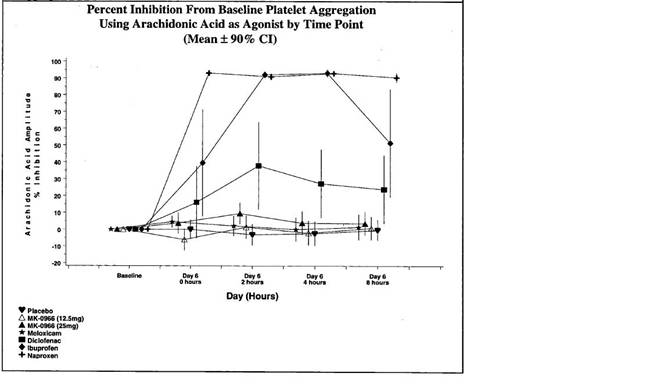
Dr Reicin explained that the graph showed that placebo, represented by the heart, had no inhibitory impact. Nor (effectively) did MK‑0966 (Vioxx), represented by the white and black triangles. Diclofenac had something of an impact, peaking at about 2 hours. Meloxicam had an impact of over 90% inhibition at the 2 and 4‑hour readings. However, naproxen, represented by the plus sign, had an effect which was both substantial and enduring. After 8 hours, the impact of naproxen was still in the order of 90% inhibition. The other test mentioned by Dr Reicin was of the ability of the various treatments to inhibit COX‑1 activity, as measured by the generation of serum thromboxane (TXB2). She said that the results showed that the weighted average inhibition of TXB2 on day 6 was about 95% for subjects taking naproxen.
92 The Protocol 061 data were very significant to Dr Reicin. They introduced some sense into what had been a somewhat confusing array of data. Dr Reicin believed that Vioxx, like placebo, had no clinically significant effect on clotting, which was borne out in the Alzheimer’s studies, where cardiovascular event rates were the same on Vioxx and placebo. Ibuprofen and diclofenac did not have a strong effect on clotting because they did not inhibit platelet clumping strongly over a sustained period of time, so it made sense to Dr Reicin that the rates of cardiovascular events were similar as between Vioxx and these NSAIDs in the Phase IIb/III osteoarthritis trials. Protocol 061 showed that naproxen was (in the words of Dr Reicin) “unique among these NSAIDs in its ability to inhibit platelets”. This, she concluded, was probably what had occurred in the VIGOR trial (although I would add that her conclusion contained the unstated assumption that the patients in the naproxen arm of the trial were highly compliant in taking their medication twice daily).
93 At 10.23 pm on Sunday 19 March 2000, Laurence Hirsch, then a member of the public affairs section at MRL, sent a draft memorandum to Dr Reicin and Dr Brian Daniels for comment. At 3.27 am on 20 March 2000, Mr Hirsch sent a further draft of that statement to Drs Reicin and Daniels, and to certain others in Merck. The memorandum was, it seems, intended for circulation within Merck subsidiaries to provide them with information about the results of VIGOR and with an understanding of Merck’s formal position in relation to those results. In his communication of 3.27 am on 20 March, Mr Hirsch said:
I think we need to seriously consider a proactive “Dear doctor” letter to US MDs, and possibly other countries as well. Logistics may be difficult, but the alternative, as discussed, of having physicians learn of this from their patients, or from the media, is not very appealing....
The draft memorandum was headed –
Standby Statement - Vioxx and Cardiovascular Events in VIGOR
Note: This information is confidential Only statements within the “shaded area” of this document are approved for use outside of Merck.
There were no “shaded” areas on the draft, but there was a box setting out an “approved public statement”. I shall refer to that shortly. The document itself opened with a paragraph headed with the word “Issue”. In the version which was sent by Mr Hirsch at 3.27 am on 20 March 2000, that paragraph was as follows:
The preliminary results of VIGOR (VIoxx Gastrointestinal Outcomes Research) indicate a significant reduction in PUBs (primary endpoint); complicated PUBs, or POBs were also reduced. There is an approximate 2‑fold increase in the risk of certain cardiovascular events, including myocardial infarctions and cerebrovascular events in these RA patients treated with Vioxx compared to naproxen. It is unclear whether these results reflect an adverse effect of rofecoxib (at the 50 mg dose) or an antiplatelet (protective) effect of naproxen, or both. Additionally, it is possible that only RA patients are at increased risk for thrombotic‑type CV AEs when treated with a coxib, as opposed to a traditional NSAID – the safety profile of Vioxx in our OA, pain, and Alzheimer’s disease studies is excellent. Due to the increase in these serious, unexpected adverse events, a number of actions need to be undertaken promptly (see below), well before reaching ‘clean file’. The implications for the arthritis & analgesia franchise are high – due to differences in study design and allowance for use of low‑dose aspirin in the celecoxib outcomes studies, there may not be similar findings in the Searle studies.
I infer that the underlined passage was inserted by way of amendment to the original version sent at 10.23 pm on 19 March.
94 There is in evidence another version of the draft memorandum, containing “tracked” changes which Dr Reicin did not deny were most likely her own (although she could not recall making them). In that version, the paragraph headed “issue” appears as follows:
The preliminary results of VIGOR (VIoxx Gastrointestinal Outcomes Research) indicate a significant reduction in PUBs (primary endpoint), and all primary and exploratory GI endpoints including– complicated PUBs, or POBs were also reduced and all GI bleeds–. There is an approximate 2‑fold increase decrease in the risk of certain cardiovascular events, including myocardial infarctions and cerebrovascular events in these RA patients treated with Vioxx naproxen compared to VIOXXnaproxen. It is unclear whether these results reflect an antiplatelet (protective) effect of naproxen, an adverse effect of rofecoxib (at the 50 mg dose) or an antiplatelet (protective) effect of naproxen, or both. If these results are due to an adverse effect of rofecoxob at the 50 mg dose, Additionally, it is possible that only RA patients are at increased risk for thrombotic‑type CV AEs when treated with a coxib, as opposed to a traditional NSAID. The incidence of thrombotic‑type CV Aes in patients treated with VIOXX was similar to placebo and the NSAID diclofenac in - the safety profile of Vioxx in our OA, pain, and Alzheimer’s disease studies is excellent. Due to the increase in these serious, unexpected adverse events, a number of actions need to be undertaken promptly (see below), well before reaching ‘clean file’. The implications for the arthritis & analgesia franchise are high – due to differences in study design, differences in the NSAID comparator and allowance for use of low‑dose aspirin in the celecoxib outcomes studies, there may not be similar findings in the Searle studies. (Note The Searle FOI reports an decreased incidence of cardiac SAEs in patients on the NSAID comparator (mainly naproxen and ibuprofen) compared to celecoxib or placebo)
One of the two main paragraphs in the box headed “Approved Public Statement” was:
Careful monitoring of adverse experiences (AEs) is an important part of all clinical trials. The preliminary analysis of cardiovascular AEs in VIGOR shows an unexpectedly (Is this unexpected? Not really we put together a cardiovascular SOP and noted in the original DAP that theoretically the naproxen rate would be lower) higher lower incidence of certain serious thrombotic (?) CV AEs in the rofecoxib naproxen group compared to the naproxen rofecoxib group. Although the data are not final, Merck has voluntarily notified the FDA and other regulatory agencies of these findings, as well as the investigators in VIGOR and in other trials of both VIOXX and MK‑663, a related investigational coxib. In addition, patients participating in ongoing studies in RA and other conditions will be receiving revised consent forms. In certain trials, the protocol will be modified to allow co‑administration of low‑dose aspirin, based on the treating physician’s recommendation. We may also send a voluntary ‘Dear Doctor’ letter to physicians In the US – TBD
TBD was, I infer, Dr Daniels. The underlined and cross‑out changes appear to have been made by Dr Reicin. The document also contained a “Questions and Answers” section, including the following:
Couldn’t this have been predicted?
We knew that there was a theoretical possibility that the antiplatelet affects of naproxen could result in a decreased incidence of these events compared to rofecoxib There was no difference in cardiovascular SAEs in previous studies in over 5000 patients with OA. This may reflect the patient population as well as the fact that the major NSAID used in the long term phase III OA studies was diclofenac which does not provide a sustained inhibition of platelet aggregation in the same way that naproxen or aspirin would. Extensive preclinical and chemical pharmacology studies indicated no change to either platelet aggregation or in bleeding time with doses of Vioxx Up to ___ There was no difference in cardiovascular SAEs in previous studies in over 5000 patients with OA.
And:
Over a year ago, scientists at the U of Pennsylvania (FitzGerald et al) published a report in PNAS suggesting there might be an increased risk of cardiovascular events in patients taking coxibs, due to changes in levels of certain prostaglandins. Were they right?
We cannot answer this with 100% certainty. The fact that the incidence of these events in patients with OA and alzheimers was similar in patients on VIOXX compared to placebo goes against this. It is however possible that patients with a tendency to hypercoaguable states (need layman wording) such as patients with Lupus or select patients with RA may be at a slightly higher risk.
Again, the changes made here appear to have been Dr Reicin’s.
95 The changes made to the “Standby Statement” by Dr Reicin lead me to infer that, by 20 March 2000 or thereabouts, she at least had become sufficiently confident that the cardiovascular results of VIGOR were to be explained by the antiplatelet effect of naproxen to advise the public affairs section that that interpretation should, in effect, be the basis of the formal position to be adopted by Merck in relation to those results. Under cross‑examination, referring to a letter sent to the FDA on 23 March 2000 (to which I shall refer shortly), Dr Reicin said:
What I would tell you is I was trying to understand the data and by the 23rd when we sent that in and over the following few months I became more and more certain that this was naproxen acting as a cardioprotective effect and not Vioxx having a prothrombotic effect. I was very, very comfortable with that as the way to understand the data.
96 On 22 March 2000 there was a teleconference involving members of the VIGOR Steering Committee. Dr Reicin participated, as did Dr Shapiro and Dr David Bjorkman (Professor of Medicine at the University of Utah School of Medicine who had been a member of the VIGOR Data Safety Monitoring Board). Some idea of the matters there discussed may be gathered from an email sent by Dr Bjorkman to Dr Shapiro on the same day:
I found the conference call very interesting, both in terms of the enormous amount of work that you have done with the data and the additional research, but also in terms of the way that the Steering Committee reacted to the news. I think that the VIGOR study unmasked the antithrombotic effects of naproxen. I think the contribution of a systemic inflammatory disorder is likely to be very small, at the most. The truth is that this IS a class effect that results from alterations in thromboxane and prostacyclin levels that are different between naproxen and rofecoxib.
I was very impressed with the way that you dealt with this issue from the ethical and safety standpoint. I think that you and Mike did a great job in pointing this issue out to the company, and I think that Merck is taking the high road that will, at least in the long run, be most profitable.
I hope that I represented the DSMB appropriately. Please let me know if I misrepresented anything so that it can be corrected. I realize that our decision to allow the trial to continue will be put under the microscope, so I want to be make sure that what I said was absolutely correct.
Notification of VIGOR results to Merck subsidiaries
97 On 22 March 2000, an email was sent from Merck to its international subsidiaries, advising them of two teleconferences to be held on 23 March (USA time). The purpose of the conferences was “to discuss new information regarding Vioxx”. The conferences were to be run by Drs Reicin and Daniels. Participants were to receive “some key slides” one hour prior to the commencement of each conference. A copy of these slides is in evidence. I should say that the exhibit to which I am about to refer was discovered by MSDA, tendered by the applicant without objection, but not referred to any of the witnesses. Dr Reicin, who was a presenter at the teleconferences, was not asked about it. Counsel for the applicant asked me to infer from its content that the exhibit represented the slides sent to international subsidiaries on 22 March 2000. They submitted that the slides were created between 9 and 23 March. Counsel for the respondents resisted the drawing of that inference. They said that a comparison of the data revealed that the slides contained different, and inferentially later, information than that sent to the FDA on 23 March. I have looked at the data in the slides, and I must say that that appears not to be so. The figures in the slides showing gastrointestinal endpoints are the same as the corresponding figures sent to the FDA on 23 March. So are the figures showing serious thromboembolic cardiovascular adverse events. This correspondence leads me to make the inference proposed by the applicant, namely, that these slides were the ones used at the international teleconferences on 23 March 2000.
98 In the slides, the Vioxx Phase III osteoarthritis trials were referred to, and it was said that the incidence of serious cardiovascular adverse events in those trials was similar as between Vioxx, NSAID comparators and placebo. The study design, objects, and patient population of the VIGOR trial were explained. The preliminary gastrointestinal results of the trial were set out, in tabular and graphical form. There followed a series of slides headed “Cardiovascular Safety”. Then came two slides as follows:
COX-1/COX-2
Effects on Platelet Aggregation
___________________________________________________________________
● Cyclooxygenase enzymes are involved in the production of both thromboxane and prostacyclin
– Thromboxane and Prostacyclin have opposing actions and normal physiology requires a balance of the two
● Thromboxane
– Platelet derived COX-1 promotes platelet aggregation via thromboxane
– The cardioprotective (ie prevention of MIs, Strokes) effects of aspirin are mediated through its complete inhibition of COX-1 mediated thromboxane formation leading to complete inhibition of platelet aggregation
– Those NSAIDs which can completely inhibit thromboxane generation and platelet aggregation could theoretically act as cardioprotective agents
– Not all NSAIDs completely inhibit platelet aggregation
– Cox-2 selective inhibitors do not inhibit platelet aggregation.
COX-1/COX-2
Effects on Platelet Aggregation
___________________________________________________________________
● COX-1/COX-2 derived prostacyclin causes blood vessels to dilate and prevents platelet aggregation
– NSAIDs, rofecoxib and celebrex result in ~50% reduction in prostacyclin levels
● Potential Implications
– NSAIDs that completely inhibit thromboxane and platelet aggregation could theoretically decrease the incidence of cardiovascular adverse events such as heart attacks and strokes compared to COX-2 selective inhibitors through its antiplatelet affects
– COX-2 specific inhibitors could theoretically cause an imbalance in the platelet aggregation through inhibition of prostacylin [sic] without inhibition of thromboxane. This imbalance could potentiate thrombotic events
99 The next slide announced that, in the VIGOR trial, “naproxen had a reduced incidence of thromboembolic cardiovascular [serious adverse experiences] compared to Vioxx”. In the Vioxx arm, it was said that the event rate was 2.12% per year, compared to 1.13% per year in the naproxen arm. Comparative figures were also given for deaths, cardiovascular deaths, myocardial infarctions, strokes, and deep vein thromboses. There was a Kaplan‑Meier plot (a means of representing statistics to which I shall refer below) of the occurrence of serious thromboembolic cardiovascular events in the two arms of the trial. The incidence of such events in the Phase III osteoarthritis trials, and in the Alzheimer’s disease studies, was set out, and was said to be “similar” in the Vioxx and placebo arms in each case. It was then pointed out that the rate of serious thromboembolic adverse experiences in the Vioxx arms of the Phase III osteoarthritis trials and in VIGOR was “similar” (said to be 2.05% per year – which I take to be per patient-year – and 2.12% per year respectively).
100 The next two slides were as follows:
What Makes VIGOR
Different?
____________________________________________________________________
● Use of Naproxen as NSAID comparator
– Like aspirin and unlike diclofenac (major NSAID comparator in Phase III studies), naproxen demonstrates sustained nearly complete inhibition of platelet aggregation
– Likely to have the cardioprotective effects seen with aspirin
● Patient population—Rheumatoid Arthritis
– Some patients with rheumatoid arthritis appear to be at particularly high risk for the development of MIs, strokes, DVTs
– Epidemiology suggest increased incidence of CV mortality in RA
– Chronic inflammation, presence of lupus anticoagulant or other clotting disorders may play a role
Cardiovascular Safety
Conclusions
____________________________________________________________________
● Naproxen demonstrated a reduced incidence of serious thromboembolic events compared to VIOXX™ due to its ability to inhibit platelet aggregation and act as a “cardioprotective agent”
– Celecoxib FOI: NSAID comparator group (mainly naproxen) had reduced rates of serious cardiac events compared to celecoxib or placebo
● The cardioprotective effects of naproxen may be enhanced in subgroups of patients rheumatoid arthritis or other inflammatory diseases who may be at an increased risk for thrombosis.
101 Next, a histogram compared the effect of Vioxx and various NSAIDs on the inhibition of platelet aggregation. Both placebo and Vioxx were shown to have little or no effect, while naproxen was shown to have, as I read it, about a 90% effect. A graph headed “COX-1 activity: TXB2 inhibition day 6” painted a similar picture.
102 The next relevant slide was as follows:
Cardiovascular Safety
Conclusions (cont)
____________________________________________________________________
● Can the increased incidence of thromboembolic events on VIOXX™ c/w naproxen be due to an adverse effect of VIOXX?
– Cannot 100% rule out but unlikely
– Incidence of CV events with VIOXX was similar to placebo in OA and Alzheimers/dementia
– Incidence of CV events with VIOXX treated patients was identical in Phase III OA and VIGOR--it was the naproxen arm which was reduced compared to placebo or diclofenac
– Any negative effect would be a class effect--ie via COX-2 inhibition of prostacyclin and likely limited to patients with an increased tendency for thrombosis
– Case reports of patients with lupus developing thrombosis soon after starting celebrex
Two slides later, and by way of conclusion, was the following:
General Conclusions
____________________________________________________________________
● VIOXX represents a new class of drugs with significantly reduced GI toxicity compared to standard NSAIDs
● VIOXX appears to have better GI tolerability compared to standard NSAIDs
● VIOXX does not provide the cardioprotective effects of some NSAIDs
● Need to be on watch for the possibility that the COX-2 inhibitor class (including VIOXX and Celebrex) may increase the risk of cardiovascular disease in patients with chronic inflammatory diseases who are at risk of thombosis [sic]
The 14‑day report to the FDA
103 The results of the VIGOR trial were the subject of a 14‑day report forwarded to the FDA on 23 March 2000. The report noted that the process of the adjudication of cardiovascular events arising in the course of the trial was, as at 9 March 2000, only about 50% complete. The data in the report were based, therefore, upon all thromboembolic cardiovascular events, regardless of the status of adjudication. They showed that, in 2736 patient‑years in the Vioxx arm, there were 58 patients who had had serious thromboembolic adverse experiences. The corresponding number in the naproxen arm (in 2735 patient‑years) was 31. The rates were 2.12 and 1.13 such events per 100 patient‑years respectively. The report expressed the relationship between these figures not as the relative risk of taking Vioxx rather than naproxen, but as the relative risk of taking naproxen rather than Vioxx. That was consistent with what had by then (although only very recently) been adopted as Merck’s formal explanation of the cardiovascular data in the trial. As expressed in the report, the relative risk of taking naproxen was 0.53 (0.35, 0.83). The difference between the two treatments was significant. Although not expressed by way of a relative risk, the report disclosed that, in the sample of 4046 patients taking Vioxx, there were 21 myocardial infarctions, while, in the sample of 4029 patients taking naproxen, there were 9 such events.
104 In the introductory section of the report, it was stated:
Platelet aggregation plays a central role in the pathophysiology underlying thromboembolic cardiovascular events such as myocardial infarction and stroke. An extensive body of research has demonstrated that therapy with aspirin, an irreversible inhibitor of COX-I and COX-2 with anti‑platelet properties, results in a reduction in the incidence of serious cardiovascular events in patients presenting with an acute coronary syndrome and in patients with history of myocardial infarction or stroke. Because COX-2 selective inhibitors do not inhibit COX-1‑mediated platelet aggregation, it was hypothesized that therapy with non-specific COX-l/COX-2 inhibitors may result in a lower incidence of serious cardiovascular events as compared to the COX-2 selective inhibitors. Such an observation would have to be factored in to the overall risk-benefit profile of the COX-2 selective inhibitors.
In the body of the report, it was stated that the VIGOR trial represented the only study to have demonstrated a difference in the incidence of adverse cardiovascular events in patients who were given rofecoxib by comparison with patients who were given either placebo or a standard NSAID. The reduced incidence of serious thromboembolic cardiovascular events in the naproxen arm in VIGOR was said to be inconsistent with previous analyses done in the Phase IIb/III osteoarthritis studies and in the Alzheimer’s studies to which I have referred.
105 For the purposes of looking at the osteoarthritis data, MRL had pooled the results of nine randomised, controlled clinical trials of rofecoxib, ranging from 6 to 86 weeks in duration, in which 5495 patients suffering from osteoarthritis participated. The overall duration of exposure was 1655 and 706 patient‑years for the combined rofecoxib (all doses) and combined NSAID (all comparators) groups, respectively. The result of this pooled analysis, according to the report of 23 March 2000, was that there were 2.05 serious thromboembolic cardiovascular adverse events for every 100 patient-years in the group taking Vioxx, and 2.27 such events per 100 patient-years in the NSAID group. The relative risk of taking an NSAID rather than Vioxx was 1.09 (0.60, 1.99). There were five myocardial infarctions in the Vioxx group (within a sample of 3357) and three in the NSAID group (within a sample of 1564).
106 Because the Alzheimer’s studies were ongoing, and staff at MRL had been only partially unblinded to the results in March 2000, the report of 23 March 2000 did not provide any information about Vioxx or placebo in terms. The treatment groups were identified as A and B (where there were 1050 patients in each group). A table indicated that there had been 26 and 32 thromboembolic cardiovascular serious adverse experiences in treatment groups A and B respectively. There had been seven myocardial infarctions in group A, and five in B.
107 The report of 23 March 2000 also dealt with another study that was then ongoing, called ADVANTAGE, the purpose of which was to evaluate the tolerability and effectiveness of 12 weeks of therapy with rofecoxib in patients with osteoarthritis of the knee, hip, hand or spine, and in which naproxen was the comparator. Patients taking low‑dose aspirin for cardioprotection were not excluded. Although the results were not final ones, the data showed no difference in the occurrence of thromboembolic cardiovascular serious adverse experiences as between the two arms (13 experiences in a population of 2795 patients in each case). There had been five myocardial infarctions in one group, and two in the other.
108 The final source of data to which MRL turned in seeking an explanation for the VIGOR results was Merck’s “Worldwide Adverse Experiences Surveillance (WAES) databases for rofecoxib”. According to the report of 23 March, this database –
… contained a total of 5576 spontaneous marketed‑use adverse experience (AE) reports. Of these 3491 were received from sources in the USA and 2085 from sources ex‑USA. Among the 5576 total reports there were 617 (11.1 %) which contained one or more AE codes mapping to the cardiovascular body system in the company’s medical codes dictionary: 431 were received from sources in the USA and 186 were received ex‑USA. Seventy‑eight of the 617 reports containing one or more cardiovascular AEs (1.4% of all reports) contained AEs designated as “thromboembolic” … Of these 47 were reported in the USA and 31 ex‑USA …
The conclusion expressed in this section of the report was that the results of VIGOR “stand in contradistinction to the cardiovascular safety findings of rofecoxib/NSAID or rofecoxib/placebo comparison studies in other populations …”
109 The report contained a lengthy section headed “Potential Explanations for the Cardiovascular Findings in the VIGOR Study”. It was said that there were two factors which, either alone or in combination, might account for the divergent cardiovascular safety findings referred to. The first factor was that NSAIDs such as naproxen might be cardioprotective by way of COX-1-mediated inhibition of platelet aggregation. It was added that VIGOR was the only trial of any length which involved patients with rheumatoid arthritis, who were known to be at higher risk of serious cardiovascular events. It was surmised that they might be more likely “to demonstrate the benefit of anti‑platelet therapy”. The second factor was, in effect, the FitzGerald hypothesis. The report noted that therapy with COX-2 selective inhibitors had been shown to cause moderate reductions in the synthesis of prostacyclin, without any COX-1 mediated inhibition of platelet aggregation. In a footnote, the report referred to McAdam (1999).
110 The report then proceeded to examine the available data with respect to these two possible explanations of VIGOR. Here, the report dealt first with the potential cardioprotective effect of COX-1 inhibition by naproxen. It discussed the role of platelets in acute coronary and cerebro‑vascular syndromes, the effects of aspirin and NSAIDs on COX‑1 activity, and what was said to be evidence to support the hypothesis that a naproxen‑mediated vascular protective effect was responsible for the reduced incidence of serious cardiovascular adverse experiences in the VIGOR trial. In the course of this discussion, the report referred to Protocol 061, to the results of which Dr Reicin had been exposed by Dr Gertz.
111 The report next referred to epidemiologic studies which had suggested that patients with rheumatoid arthritis had an increased incidence of cardiovascular disease. It was said that the etiology of the increased risk was uncertain, but might have related to increased atherogenesis in the setting of chronic inflammation. The report noted that the presence of anticardiolipin (aCL) antibodies (associated with vascular thrombosis) was “classically associated with systemic lupus erythematosus (SLE)”, and said that aCL antibodies had been demonstrated in 16% to 33% of patients with rheumatoid arthritis. After some discussion of these aspects, the report continued:
However, a subgroup of patients with [rheumatoid arthritis], particularly those with detectable aCL antibody titers, appear to be at particular risk of sustaining a thrombotic cardiovascular event.
The etiology of this increased tendency for thrombosis in patients with [rheumatoid arthritis] or detectable aCL antibodies is not well understood. … aCL‑positive patients with lupus and without lupus have been shown to have a 4 fold increase in TXA2 synthesis as measured by urinary excretion of TXB‑M as well as a small increase in prostacyclin synthesis as measured by prostacyclin urinary metabolites as compared to healthy age matched controls. Treatment with low‑dose aspirin significantly reduced urinary TXB‑M but did not significantly affect levels of prostacyclin metabolites … These findings suggest that patients with [rheumatoid arthritis] (or a subgroup) are at increased risk of having a baseline hypercoagulable state. … Thus, the cardioprotective effects of TXA2 inhibition could be exaggerated in the rheumatoid patient population and may also explain the discrepant cardiovascular safety results of VIGOR versus studies in patients with osteoarthritis and Alzheimer’s Disease. Although the aggregate of clinical and postmarketing data does not suggest that rofecoxib increases the risk of patients sustaining a thromboembolic event, the negative impact of COX‑2 selective inhibitors such as rofecoxib in a small subgroup of patients with [rheumatoid arthritis] or other chronic inflammatory diseases cannot be ruled out. Inhibition of prostacyclin synthesis as measured by urinary PGI‑M has been shown to be reduced by both nonselective COX‑1/COX‑2 inhibitors as well as by the selective inhibitors of COX‑2, rofecoxib and celecoxib … COX‑2 selective inhibitors, therefore have the potential to create an imbalance in the coagulation cascade through inhibition of prostacylin without thromboxane inhibition. If this partial inhibition of prostacyclin is of clinical relevance, it may be limited to a subset of susceptible patients. Based on the finding that serious thromboembolic events were not increased compared with placebo in patients with dementia or [osteoarthritis], it is likely that in normal patients, a 50% decrease in prostacyclin without TXA2 inhibition is without clinical consequence. However, patients with a hypercoagulable state such as [rheumatoid arthritis] or Lupus patients who are aCL‑positive patients and have decreased levels of protein S might be particularly susceptible to this partial inhibition. Of note, there have been case reports of 2 patients with SLE, high levels of aCL antibodies and low levels of protein S who sustained thrombotic events shortly after the initiation of celecoxib … However these events occurred within a short time after the initiation of celecoxib therapy. The difference observed cardiovascular events in VIGOR was not apparent until 6 weeks after initiation of study therapy. This implies that an acute imbalance of prostacyclin and thromboxane did not account for the findings in VIGOR.
112 The two patients referred to towards the end of this extract were two of the three mentioned by Dr Oates in his letter of 13 August 1999 and which were the subject of the paper at the American College of Rheumatology proceedings to which I have referred. In her oral evidence, Dr Reicin explained what she did by way of inquiry into the possibility of SLE cases having some relevance to the results of VIGOR. She said:
I embarked on a literature search to see if rheumatoid arthritis patients had ever been reported to have similar type of prothrombotic tendencies as patients with lupus or SLE, so wondering if what we had seen in VIGOR may have been somehow related to that, and I found some obscure literature on it and proposed a hypothesis that maybe rheumatoid arthritis patients were somehow similar to patients with lupus. I am not a rheumatologist by training and when I discussed that proposal with rheumatologists, both in and outside of Merck, I didn’t gain a lot of enthusiasm for what I was proposing. I did … however, go through some of that literature … in the March 23rd document that was sent down to the FDA.
113 The summary at the end of the report included in the following passage:
An extensive review of rofecoxib clinical trials databases and post marketing surveillance does not suggest that rofecoxib causes an increased rate of serious thromboembolic cardiovascular events. The similar incidence of these events in patients treated with rofecoxib, placebo and the NSAID diclofenac (which does not provide complete and sustainable inhibition of platelet aggregation) as well as post marketing surveillance studies supports this conclusion. The reduced incidence of serious thromboembolic cardiovascular events in patients with rheumatoid arthritis treated with naproxen compared to rofecoxib is likely the result of the cardioprotective effects of naproxen, a potent inhibitor of platelet aggregation. It is possible that these effects may be enhanced in a small subgroup of patients with rheumatoid arthritis or other inflammatory diseases who may have an increased tendency to the development of pro‑thrombotic states. Although the role of COX‑2 specific inhibitors such as rofecoxib in potentiating a pro‑thrombotic state in patients with rheumatoid arthritis cannot be ruled out, this scenario seems less likely. In clinical studies of patients with OA or memory impairment, this potential alteration in the balance between thromboxane and prostacyclin was of no apparent clinical consequence.
Ongoing consideration of VIGOR results
114 Returning to the narrative, on 24 March 2000, Dr Nies forwarded to Dr Reicin an email which he had just received from Dr FitzGerald. It referred to an article which was then in press and which was published later that year as Rodriguez (2000). Dr FitzGerald said that he had emailed the leading author of the article, and proceeded to set out figures which do not appear to be derived from the article itself, and which, I infer, were obtained by Dr FitzGerald by way of email. In his email to Dr Nies, Dr FitzGerald said:
[aspirin] significantly reduced the risk of first nonfatal [myocardial infarction] in these females rr 0.56 (0.26 – 1.21)
all NSAIDs had no effect 1.32 (0.97 – 1.81)
individual nsaids had no significant effect on either fatal, nonfatal or total [myocardial infarctions].
amongst these INSIGNIFICANT effects, naproxen looked best 0.33 (0.07 – 1.4) but was closely followed by ibuprofen 0.57 (0.2 – 1.6). Diclofenac tended to be worse 1.66 (0.99 – 2.76). There were no sig diffs between the nsaids.
this is the best comparative clin data on [myocardial infarction] and NSAlDs that I am aware of.
Dr FitzGerald’s email was put to Dr Reicin under cross‑examination, with the suggestion, implicitly, that she should have regarded it as counting against the plausibility of her conclusion that the myocardial infarction results in VIGOR should be explained by the beneficial effect of naproxen. Dr Reicin said that, other than to note that it was consistent with her thinking about naproxen, the email did not influence her. She pointed to the contradiction between Dr FitzGerald’s statement that the risk reduction with aspirin was significant and the fact that the confidence interval straddled unity. She noted the non‑significant relative risk of taking naproxen, shown to be 0.33 (point estimate).
115 On 27 March 2000, Merck issued a media release which announced the favourable gastrointestinal results of the VIGOR trial and continued:
In addition, significantly fewer thromboembolic events were observed in patients taking naproxen in this GI outcomes study, which is consistent with naproxen’s ability to block platelet aggregation. This effect on these events had not been observed previously in any clinical studies for naproxen. Vioxx, like all COX-2 selective medicines, does not block platelet aggregation and therefore would not be expected to have similar effects. As a result, Merck is notifying investigators, who are conducting other Merck studies with Vioxx or another investigational medicine in the same class, of protocol amendments to allow the addition of low‑dose aspirin where appropriate. Patients using low‑dose aspirin, which also blocks platelet aggregation, were excluded from the GI outcomes study. Vioxx does not interfere with the ability of low‑dose aspirin to block platelet aggregation.
…
Researchers believe that NSAIDs work by inhibiting two related enzymes: COX-1, the enzyme that helps maintain the stomach lining and promotes platelet aggregation, and COX-2, the enzyme that triggers pain and inflammation. At therapeutic doses, Vioxx works by selectively inhibiting COX-2 without inhibiting COX-1; non‑selective NSAIDs like naproxen inhibit both COX-1 and COX-2. Medicines like aspirin and naproxen that significantly inhibit COX-1 block platelet aggregation and therefore have the potential to provide cardioprotection.
116 One of the authors of Rodriguez (2000) was Prof Carlo Patrono of the Department of Medicine and Ageing, Universiti di Chieti “G. d’Annunzio”, Chieti, Italy. On 28 March 2000, an employee of Merck sent an email to Dr Reicin and others in which he recounted a conversation held with Prof Patrono on 25 March 2000. Prof Patrono had expressed the view that the difference between the arms in the VIGOR trial could not be explained by the cardioprotective effect of naproxen, and most likely was the result of chance. Prof Patrono confirmed those views in an email to the employee on 3 April 2000. For her own part, Dr Reicin never “gave strong consideration” to the possibility that the results arose by chance. She accepted the statisticians’ analysis that the 95% confidence interval with respect to the relative risk of taking naproxen lay wholly below unity. When she was informed of Prof Patrono’s views, she said (in an email to Dr Nies) “I think we should talk directly to Carlo”. Dr Reicin gave evidence that, over a long period, she had had conversations with Prof Patrono about the VIGOR results. Initially, it had been his view that one would not get sustained inhibition of TXB2 from taking naproxen, but, according to Dr Reicin, he later did his own work on the subject and came to the view that naproxen was capable of explaining at least part of the results of VIGOR, with the other part being a matter of chance.
117 On 12 April 2000, Dr Scolnick sent Dr Reicin an email in which he said:
I will tell you my worry quotient is high. I actually am in minor agony. What I really want to do is a 10000 vs 10000 patient study in mild‑moderate [osteoarthritis] Tylenol vs vioxx with prn low dose [aspirin] for those judged to need it. Safety first primary endpoint and effiacy secondary or co primary. WE WILL NOT KNOW FOR SURE WHAT IS GOING ON UNTIL WE DO THIS STUDY. PLEASE THINK HARD ABOUT THE DESIGN BEFORE THE PAC MEETING.
Under cross‑examination, Dr Reicin was asked whether she took this email as an expression of doubt on the part of Dr Scolnick as to whether the naproxen theory adequately explained the VIGOR results. She responded that she could not tell whether that was what Dr Scolnick was thinking. Pressed to say whether she had ever asked Dr Scolnick why he was so worried, Dr Reicin said:
I did not ask him that. I understood that as head of Merck Research Laboratories he would be worried about the safety of our medicines. That was his job.
However, Dr Reicin did reply to Dr Scolnick’s email, in the following terms:
I have a lot of concerns about design issues for this type of trial. We have a meeting set up with you for next tuesday afternoon to review some of the [cardiovascular] data, review plans for exploring other databases etc. I will try to sit down with one of the statisticians before then and come up with some assumptions and harder numbers.
What, if anything, transpired from the proposed meeting referred to by Dr Reicin, or from her conversations with the statisticians, was not the subject of evidence.
118 On 14 April 2000 Merck notified the FDA of an amendment to one of the protocols for the Alzheimer’s trials (which, as I have said, were ongoing). In a number of places, the sentence “Patients who develop a need for cardio‑protective doses of aspirin after randomization are permitted to use aspirin ≤100 mg/day” was added. Similar notifications were given with respect to two other protocols for these trials on 25 and 26 April 2000. I would infer that these amendments were made to allow the use of a conventional antiplatelet agent for patients taking Vioxx.
119 In May 2000, the chair of the VIGOR Steering Committee, Dr Claire Bombardier, and others, including Dr Reicin, submitted the VIGOR results for publication to the New England Journal of Medicine. Although not published until November 2000, (Bombardier 2000), this article demonstrated the way Dr Reicin and her colleagues were thinking in May of that year. Conformably with its title, the article concentrated upon the gastrointestinal dimension of the VIGOR results. But it presented also the cardiovascular results, in terms of which the applicant was critical in the present proceeding.
120 In that part of the article that set out the design of the study in VIGOR, the authors stated:
Because highly selective cyclooxygenase‑2 inhibitors do not inhibit platelet aggregation, which is mediated by cyclooxygenase‑1, there was a possibility that the incidence of thrombotic cardiovascular events would be lower among patients treated with nonselective cyclooxygenase inhibitors than among those treated with cyclooxygenase‑2‑selective inhibitors. Therefore, cardiovascular events were also assessed for a future meta‑analysis by independent committees whose members were unaware of the patients’ treatment assignments. A separate analysis of these events, however, was not specified in the study design.
Under the subheading “General Safety” in the section of the article which set out the results of the trial, the authors stated:
The safety of both rofecoxib and naproxen was similar to that reported in previous studies. The mortality rate was 0.5 percent in the rofecoxib group and 0.4 percent in the naproxen group. The rate of death from cardiovascular causes was 0.2 percent in both groups. Ischemic cerebrovascular events occurs in 0.2 percent of the patients in each group. Myocardial infarctions were less common in the naproxen group than in the rofecoxib group (0.1 percent vs. 0.4 percent; 95 percent confidence interval for the difference, 0.1 to 0.6 percent; relative risk, 0.2; 95 percent confidence interval, 0.1 to 0.7). Four percent of the study subjects met the criteria of the Food and Drug Administration (FDA) for the use of aspirin for secondary cardiovascular prophylaxis (presence of a history of myocardial infarction, angina, cerebrovascular accident, transient ischemic attack, angioplasty, or coronary bypass) but were not taking low‑dose aspirin therapy. These patients accounted for 38 percent of the patients in the study who had myocardial infarctions. In the other patients the difference in the rate of myocardial infarction between groups was not significant (0.2 percent in the rofecoxib group and 0.1 percent in the naproxen group). When the data showing a reduction in the rate of myocardial infarction in the naproxen group became available after the completion of this trial, Merck, the manufacturer of rofecoxib, notified all investigators in ongoing studies of a change in the exclusion criteria to allow patents to use low‑dose aspirin. There was no association between hypertension and myocardial infarction; only a single patient (in the rofecoxib group) had both hypertension and a myocardial infarction as adverse events.
Counsel for the applicant criticised this passage for having in effect “flipped” the results of VIGOR, to express them as though the trial had shown the beneficial effect of naproxen, rather than the harmful effect of Vioxx. They also noted that the article took no account of events which had been reported after the VIGOR cut‑off date and which were, it seems, known to at least some of the authors by July 2000. That aspect was the subject of a controversy between the editors of the New England Journal of Medicine and the authors of the article in 2005 and 2006, and I refer to it below.
The VIGOR Clinical Study Report
121 On 29 June 2000, MRL submitted a supplementary New Drug Application to the FDA. It incorporated the Clinical Study Report for the VIGOR trial, a 100‑page appendix to which was an analysis (dated 21 June 2000) of the cardiovascular events in the trial. The Executive Summary of that analysis contained the following statement:
The VIGOR study showed that therapy with rofecoxib was associated with a marked reduction in the incidences of gastroduodenal perforations, symptomatic peptic ulcers, and GI bleeds (PUBs) compared with naproxen therapy. On the other hand, therapy with naproxen was associated with a 56% reduction in the risk of confirmed thrombotic cardiovascular serious adverse experiences relative to therapy with rofecoxib. The reduction in cardiovascular events in general and MI in particular was accentuated in the 4% of patients in the study who were enrolled in violation of the protocol exclusion criteria because they had an indication for chronic aspirin therapy (i.e., a history of symptomatic coronary and cerebrovascular disease) but who were not receiving aspirin or other antiplatelet therapy. The magnitude of the reduction in cardiovascular events with naproxen therapy relative to rofecoxib therapy in patients without a clear indication for aspirin therapy was substantially smaller.
122 By the time of this analysis, the process of adjudication of cardiovascular events had reached the stage where it was possible for confirmed events in the VIGOR trial to be reported. In the Vioxx arm, there were now said to be 1.52 confirmed thrombotic events per 100 patient years. The corresponding figure for the naproxen arm was 0.67. The relative risk of taking naproxen rather than Vioxx, as expressed in the Clinical Study Report, was, therefore, 0.44 (0.25, 0.76). This was the figure that underlay the statement in the report that therapy with naproxen was associated with a 56% reduction in risk. The analysis dealt also with the relative risk of experiencing a myocardial infarction, as demonstrated in the trial. Here there were 17 events (0.63 events per 100 patient years) in the Vioxx arm, and 4 events (0.15 events per 100 patient years) in the naproxen arm. The relative risk again favoured naproxen: 0.24 (0.08 – 0.70).
123 Another aspect of the VIGOR results mentioned in the analysis related to the circumstance that, “in violation of inclusion criteria”, 4% of the patients in the overall cohort were indicated for the regular taking of low‑dose aspirin for cardioprotection. This meant that these patients had medical histories involving cerebrovascular accident, transient ischaemic attack, myocardial infarction, unstable angina, stable angina, coronary artery bypass graft surgery, or percutaneous coronary intervention. They were, conformably with the design of the study, precluded from taking aspirin during the trial, but it seems that they should have been. A disproportionate number of the adverse events were experienced by these patients. For example, some 38% of the myocardial infarctions were suffered within this 4% of the group. Of the 17 myocardial infarctions in the Vioxx arm, 8 were suffered by patients in this group. Of the 4 myocardial infarctions in the naproxen arm, none were suffered by patients in this arm. The report described these results as follows:
These data show that a disproportionate amount of the benefit observed with naproxen therapy in the VIGOR study occurred in patients in whom potent antiplatelet therapy was already indicated. The findings in the VIGOR study reinforce the benefit of aspirin therapy in patients at risk for thrombotic cardiovascular events. Rofecoxib does not have antiplatelet properties and therefore cannot substitute for aspirin. Thus, in patients with an indication for therapy with rofecoxib who are also at increased cardiovascular risk, concomitant therapy with aspirin and rofecoxib should be considered.
124 In the analysis, the following summary was provided of the cardiovascular outcomes of the VIGOR trial:
1. The primary objective of the VIGOR study was to determine whether chronic therapy with the highest approved dose of rofecoxib (twice the highest dose approved for chronic use), a COX‑2 selective inhibitor, would result in significant reductions in the incidence of PUBs relative to therapy with standard doses of naproxen, a potent nonselective COX‑l/COX‑2 inhibitor.
2. To ensure that the primary objective of the study was clearly addressed, non‑protocol use of COX‑l/COX‑2 inhibitors, including aspirin, was prohibited.
3. In the VIGOR study population, therapy with naproxen was associated with a reduction in the incidence of thrombotic cardiovascular serious adverse experiences. The difference between the treatment groups in the rates of thrombotic cardiovascular events was due primarily to a reduction in the incidence of myocardial infarction in patients treated with naproxen.
4. A disproportionate number of thrombotic cardiovascular serious adverse experiences occurred in the small group of study patients in whom vascular‑protective antiplatelet therapy was clearly indicated. Furthermore, the reduction in the risk of a serious thrombotic cardiovascular event that was observed with naproxen therapy in the VIGOR study was due disproportionately to a substantial reduction in such events associated with naproxen relative to rofecoxib in this small patient population. The benefit of naproxen relative to rofecoxib in the larger patient population that was not eligible for vascular‑protective antiplatelet therapy was of smaller magnitude.
5. Naproxen’s potent antiplatelet effects were evident in the VIGOR trial. Patients who received naproxen experienced a significantly higher rate of bleeding events typically associated with high‑level platelet inhibition than patients who received rofecoxib, which has no antiplatelet effects.
6. The findings in the VIGOR study reinforce the benefit of aspirin therapy in patients at risk for thrombotic cardiovascular events. Because rofecoxib does not inhibit COX-l at any dose, it does not have antiplatelet properties and therefore cannot substitute for aspirin. Thus, in patients who are being treated with COX-2 selective inhibitors such as rofecoxib and who have an indication for vascular‑protective aspirin therapy due to an increased cardiovascular risk, concomitant therapy with aspirin and COX-2 selective inhibitors should be considered.
125 The analysis dealt with the results (to the extent that they were available) of completed and ongoing studies of Vioxx in populations of rheumatoid arthritis (other than VIGOR), osteoarthritis and Alzheimer’s patients. It was said that “an extensive review of cardiovascular safety data from completed and ongoing clinical trials of rofecoxib, as well as the postmarketing experience with rofecoxib, was performed.” There were three studies to evaluate the efficacy and safety of Vioxx in patients with rheumatoid arthritis (Protocols 068, 096, and 097). Protocol 068 had been unblinded, and the unadjudicated thrombotic cardiovascular serious adverse experiences recorded in it were set out in a table. For patients taking Vioxx 25 mg daily, the event rate per 100 patient‑years was 0.07%. There were no events in the placebo group. The overall numbers appear to be quite small in both cases. Protocols 096 and 097 remained blinded, and little if anything of value was said about them.
126 The events recorded in the osteoarthritis Phase IIb/III trials were also referred to. To the extent that they involved a comparison with an NSAID, I have referred to them in para 105 above. There were also comparisons with placebo that were dealt with in this analysis: with respect to unadjudicated thrombotic cardiovascular serious adverse experiences, the relative risk of taking Vioxx rather than placebo was 1.05 within wide confidence intervals of 0.27 – 4.02. Taking all the osteoarthritis data into account, the author of the analysis said:
Thus, in evaluating the osteoarthritis trials and the VIGOR study in aggregate, the only outlier was the thrombotic cardiovascular serious adverse experience event rate in the naproxen arm of the VIGOR study, which was substantially lower than the event rates observed in the various treatment arms of the osteoarthritis Phase lIb/III database or the rofecoxib arm of the VIGOR study.
127 The analysis dealt also with the Alzheimer’s studies and with Merck’s post‑marketing experience, to each of which I have referred above in the context of the report of 23 March 2000. According to the analysis, these data too failed to show the elevated risk of taking Vioxx which emerged from the VIGOR data. In summary, the author of the analysis provided the following conclusions about the data generally:
1. In the VIGOR study, rheumatoid arthritis patients who received naproxen experienced a lower rate of thrombotic cardiovascular serious adverse experiences compared with patients who received rofecoxib.
● This imbalance was due in large part to a reduction in the incidence of myocardial infarction in the small cohort of patients enrolled in the study in violation of the protocol due to an indication for aspirin therapy on the basis of a documented history of symptomatic coronary artery or cerebrovascular disease.
● In the larger patient cohort that did not have a history of such diseases, the difference in the incidence of thrombotic cardiovascular serious adverse experiences between therapy with naproxen and rofecoxib was smaller.
2. The results of the VIGOR study stand in contradistinction to the cardiovascular safety findings of rofecoxib/nonselective NSAID or rofecoxib/placebo comparison studies in other populations (osteoarthritis, early Alzheimer’s disease). In these studies, the incidences of serious thrombotic cardiovascular serious adverse experiences were comparable between patients who received rofecoxib, placebo, or comparator nonselective NSAIDs.
3. Postmarketing surveillance has indicated a low incidence of spontaneous reports of thrombotic cardiovascular adverse experiences. This reporting rate is comparable to that observed in postmarketing surveillance of other noncardiovascular drugs.
128 Noting that VIGOR was the only study to have demonstrated a difference in the instance of thrombotic cardiovascular serious adverse experiences as between patients treated with Vioxx and those treated otherwise, the analysis turned to consider the factors, either alone or in combination, that might account for the results seen in VIGOR. Two possibilities were proposed: first, the “vascular‑protective effects of potent non‑selective COX-1/COX-2/inhibitor[s] such as naproxen relative to selective COX-2 inhibitors such as celecoxib and rofecoxib”, and, secondly, “increased risk of thrombotic cardiovascular disease in rheumatoid arthritis patients”. The analysis dealt with each of those possibilities in turn. With respect to the first, it was organised according to the following subheadings:
- The Role of Platelets in Acute Coronary and Cerebrovascular Syndromes
- The Mechanism of Action for the Vascular‑Protective Properties of Aspirin
- Comparison of the Effects of COX-2 Selective Inhibitors and Nonselective NSAIDs/Aspirin on COX-1‑Related Platelet Metabolism
- Evidence of the Antiplatelet Effect of Naproxen in the VIGOR Study
- Clinical Trials Evaluating the Vascular‑Protective Properties of Nonselective NSAIDs
- Evidence to Support the Hypothesis That a Naproxen‑Mediated Vascular‑Protective Effect was Responsible for the Reduced Incidence of Thrombotic Cardiovascular Serious Adverse Experiences in the VIGOR Study
- Evidence Against a Role for the Prostacyclin Inhibition Observed With Rofecoxib (and Other COX-2 Selective Inhibitors) in the Cardiovascular Results of the VIGOR Study
The analysis contained the conclusion, expressed on the basis of various studies, that naproxen was one of a limited number of NSAIDs that were potent in their antiplatelet effects and tended to be associated with clinical outcomes involving bleeding. A reference was made to Protocol 023, to the corresponding study involving celecoxib, and to the FitzGerald hypothesis which resulted from them. As to whether that might explain the results in VIGOR, it was stated:
The reduction in PG2 levels without reductions in TXA2 observed with both COX-2 selective inhibitors could theoretically lead to a mild imbalance in the overall control of platelet aggregation, and a mild proaggregatory state in patients receiving these compounds versus patients receiving nonselective COX‑l/COX‑2 inhibitors.
…
Taken together, the data from the celecoxib and rofecoxib studies demonstrate that therapy with these compounds is not accompanied by the antiplatelet effects observed with potent nonselective COX-l/COX-2 inhibitors such as aspirin and naproxen. Thus, COX-2 selective compounds such as celecoxib and rofecoxib, which do not affect TXA2 metabolism, platelet aggregation and bleeding times would not be expected to provide vascular‑protective effects similar to those observed following chronic aspirin therapy. The moderate reductions in the synthesis of prostacylin without COX-1 mediated inhibition of platelet aggregation … observed with COX-2 selective inhibitors theoretically could have mildly proaggregatory platelet effects. The subclass of nonselective COX-l/COX-2 inhibitor NSAIDs (such as diclofenac), which lack sustained inhibition of platelet TXA2, cause modest platelet aggregation inhibition, and do not affect bleeding time, are also not likely to have vascular‑protective properties comparable to those of aspirin … On the other hand, the subclass of nonselective NSAIDs (such as naproxen) capable of potent, sustained COX-l/COX-2 inhibition, maximal inhibition of TXA2 synthesis and platelet aggregation, and prolonged bleeding times are likely to provide a vascular protection efficacy profile similar to that of aspirin …
Nonetheless, it was stated that the “mild pro‑aggregatory state in some patients due to the partial inhibition of prostacyclin synthesis” was a less likely explanation for the results of VIGOR than the potent anti‑aggregatory effect of naproxen which, according to the analysis, had been observed in a number of studies.
129 With respect to the second possibility, the existence of epidemiologic studies which suggested that patients with rheumatoid arthritis had an increased incidence of cardiovascular disease was noted. It was said that the etiology of this, while uncertain, might relate to increased atherogenesis in the setting of chronic inflammation, accompanied by the presence of circulating prothrombotic factors. Reference was made to Ridker (2000), in which the researchers examined the effect of serum C‑reactive protein levels, a marker of ongoing inflammation, on cardiovascular mortality in an otherwise healthy population of women enrolled in the Women’s Health Study. It was said that the risk for cardiovascular morbid events increased progressively with higher levels of C‑reactive protein. It was noted that similar results had been observed in men (in which respect Ridker (1998) was referred to). It was noted that inflammation appeared to be an independent contributing factor to the excess cardiovascular mortality observed in a rheumatoid arthritis database called ARAMIS. It was proposed that the presence of increased circulating levels of prothrombotic factors may also contribute to the increased cardiovascular risk associated with rheumatoid arthritis. It was said that the presence of anticardiolipin (aCL) antibodies, had been demonstrated in 16%‑33% of patients with rheumatoid arthritis, and that recent reports had confirmed that increased levels of aCL antibodies, in some patients with rheumatoid arthritis, resulted in an increased risk of arterial or venous thrombosis. Studies were referred to in this regard. It was said that the etiology of the increased tendency for thrombosis in patients with rheumatoid arthritis or detectable aCL antibodies was not well‑understood. The text continued:
... aCL‑positive patients with and without SLE have been shown to have a 4‑fold increase in TXA2 synthesis as measured by urinary excretion of TXB‑M as well as a small increase in PGI2 synthesis as measured by PGI2 urinary metabolites compared with healthy age matched controls … Treatment with low‑dose aspirin significantly reduced urinary TXB‑M but did not significantly affect levels of PGI2 metabolites … These findings suggest that patients with rheumatoid arthritis (or a subgroup of these patients) are at increased risk of having a hypercoagulable state. Inhibition of TXA2 by aspirin or potent COX-l inhibiting nonselective NSAIDs may decrease the incidence of thrombotic events in the subset of patients at risk. Of note, aspirin has been shown to significantly decrease the incidence of recurrent thrombosis and spontaneous abortions in aCL‑positive patients … Thus, the cardioprotective effects of TXA2 inhibition could be exaggerated in the rheumatoid patient population, particularly in patients with a past medical history of symptomatic cardiovascular disease who are at high risk for spontaneous atherosclerotic plaque rupture and platelet‑mediated arterial thrombosis. This observation may have contributed to the discrepant cardiovascular results in the VIGOR study relative to the studies in osteoarthritis and Alzheimer’s disease patients. Although the aggregate of clinical and postmarketing data does not suggest that rofecoxib increases the risk of patients sustaining a thrombotic event, the negative impact of COX-2 selective inhibitors such as rofecoxib or celecoxib in a small subgroup of patients with rheumatoid arthritis or other chronic inflammatory diseases cannot be definitively ruled out. PGI2 synthesis has been shown to be reduced by both nonselective COX-l/COX-2 inhibitors as well as by the selective inhibitors of COX-2, rofecoxib and celecoxib … COX-2 selective inhibitors, therefore, have the potential to create an imbalance in platelet function through inhibition of prostacylin without thromboxane inhibition. If this partial inhibition of PGI2 is of clinical relevance, it may be limited to a subset of susceptible patients.
130 In summary, it was stated in the analysis that the results of VIGOR most likely arose from “a convergence of several protocol design factors, all serving to uniquely accentuate the cardioprotective effects of naproxen relative to rofecoxib”. The specific design factors mentioned were the use of naproxen, “a potent anti‑platelet drug”, and the nature of the study population, rheumatoid arthritis patients who “may have included a sub‑group of patients who derived particular benefit from anti‑platelet therapy with naproxen”. It was said that –
… [i]n patients at low risk for cardiovascular disease due to the absence of symptomatic atherosclerotic cardiovascular disease or absence of a chronic inflammatory, prothrombotic state, therapy with rofecoxib and nonselective NSAIDs is accompanied by similar rates of thrombotic cardiovascular serious adverse experiences …
Finally, with respect to the FitzGerald hypothesis, the conclusion of the analysis was as follows:
The effect of rofecoxib on prostacyclin synthesis is not likely to have contributed to the cardiovascular findings of the VIGOR study. Therapy with rofecoxib, placebo, and the nonselective NSAID diclofenac (which does not provide complete and sustainable inhibition of platelet aggregation) resulted in similar rates of cardiovascular thrombotic serious adverse experiences. In addition, postmarketing surveillance has reported a rate of spontaneous cardiovascular adverse reporting comparable to that observed with other noncardiovascular drugs administered to similar patient populations.
The Safety Update Report of 13 October 2000
131 On 13 October 2000, MRL provided a Safety Update Report to the FDA with respect to the cardiovascular safety aspects of the VIGOR trial. The original data had been based on a cut‑off date of 10 February 2000, since when there had been a further 11 patients who had experienced cardiovascular serious adverse experiences eligible for adjudication. Since this report contained what was effectively the final set of cardiovascular safety data from the VIGOR trial, it is appropriate to set out a number of tables that appeared in it. As in all representations of data by Merck at this time, the relative risk figures represent the risk of taking naproxen rather than Vioxx.
132 The summary of the CVT serious adverse experiences occurring in the VIGOR trial was as follows:
Summary of Analysis of Confirmed Adjudicated Thrombotic Cardiovascular Serious
Adverse Experiences VIGOR Study in Patients With Rheumatoid Arthritis
Updated Application Data
|
Event Category |
Treatment Group |
N |
Patients With Events |
PYR† |
Rates‡ |
Relative Risk§ |
||
|
Estimate |
95%CI |
|||||||
|
All thrombotic events |
Rofecoxib Naproxen |
4047 4029 |
45 19 |
2697 2698 |
1.67 0.70 |
0.42 |
(0.25, 0.72) |
|
|
All cardiac events |
Rofecoxib Naproxen |
4047 4029 |
28 10 |
2698 2698 |
1.04 0.37 |
0.36 |
(0.17, 0.74) |
|
|
All cerebrovascular events |
Rofecoxib Naproxen |
4047 4029 |
11 8 |
2699 2699 |
0.41 0.30 |
0.73 |
(0.29, 1.80) |
|
|
All peripheral vascular events |
Rofecoxib Naproxen |
4047 4029 |
6 1 |
2699 2699 |
0.22 0.04 |
0.17 |
(0.00, 1.37) |
|
|
In keeping with the data analysis section of the Adjudication SOP, this table does not include events determined by adjudications to be hemorrhagic cerebrovascular accidents. † Per 100 patient‑years at risk (PYR) § Relative risk of naproxen with respect to rofecoxib from unstratified Cox model where the number of cases is at least 11, otherwise relative risk is ratio of rates. Although a patient may have had 2 or more serious adverse experiences, the patient is counted only once within a category. The same patient may appear in different categories. |
||||||||
133 The report included a Kaplan‑Meier plot of the cardiovascular serious adverse experiences confirmed in the VIGOR. That is a plot of the cumulative build‑up in the rate (%) of experiences over time. The plot was as follows:
134 The CVT serious adverse experiences were broken down according to whether the patients concerned were, or were not, aspirin‑indicated:
Incidence of Adjudicated Thrombotic Cardiovascular Serious Adverse Experiences in Patient Subgroups
Based on a Past Medical history Meeting Criteria for Vascular protective Aspirin Therapy
VIGOR Study in Rheumatoid Arthritis Patients
Updated Application Data
|
Subgroup |
Treatment Group |
N |
Patients With Events |
PYR† |
Rates‡ |
Relative Risk§ |
||
|
Estimate |
95%CI |
|||||||
|
All patients |
Rofecoxib Naproxen |
4047 4029 |
45 19 |
2697 2698 |
1.67 0.70 |
0.42 |
(0.25, 0.72) |
|
|
Aspirin indicated ¶ |
Rofecoxib Naproxen |
170 1519 |
15 3 |
105 102 |
14.29 2.94 |
0.20 |
(0.06, 0.71) |
|
|
Aspirin not indicated║ |
Rofecoxib Naproxen |
3877 3878 |
30 16 |
2592 2596 |
1.16 0.62 |
0.53 |
(0.29, 0.97) |
|
|
† Patient‑years at risk. ‡ Per 100 PYR. § Relative risk of naproxen with respect to rofecoxib from unstratified Cox model where the number of cases is at least 11, otherwise relative risk is ratio of rates. ║ The “Aspirin Indicated” cohort represents those patients with a past medical history of cerebrovascular accident, transient ischemic attack, myocardial infarction, unstable angina, stable angina, coronary artery bypass graft surgery, or percutaneous coronary intervension. The “Aspirin Not Indicated” cohort represents those patients who did not have a past medical history of any of these diseases. ¶ Treatment by aspirin indicated subgroup interaction test, p=0.177 |
||||||||
135 The report also divided up the cardiovascular events by category and by reference to whether patients were, or were not, aspirin‑indicated:
|
Event Category |
Treatment Group |
N |
Number of Patients with Events |
PYR† |
Rates‡ |
Relative Risk§ |
||
|
Estimate |
95%CI |
|||||||
|
All Patients |
||||||||
|
Cardiovascular deaths║, MI, CVA
Cardiovascular deaths║
MI
Stroke¶ |
Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen |
4047 4029 4047 4029 4047 4029 4047 4029 |
35 18 7 7 20 4 11 9 |
2698 2698 2700 2699 2699 2699 2699 2699 |
1.30 0.67 0.26 0.26 0.74 0.15 0.41 0.33 |
0.51
1.00
0.20
0.82 |
(0.29, 0.91)
(0.07, 0.58)
(0.34, 1.97) |
|
|
Aspirin Indicated |
||||||||
|
Cardiovascular deaths║, MI, CVA
Cardiovascular deaths║
MI
Stroke¶ |
Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen |
170 151 170 151 170 151 170 151 |
12 3 1 2 8 0 3 2 |
105 102 106 102 105 102 106 102 |
11.42 2.94 0.95 1.96 7.60 0.00 2.84 1.96 |
0.26
2.07
0.00
0.69 |
(0.07, 0.91)
(0.11,122.10)
(0.00, 0.60)
(0.06, 6.02) |
|
|
Aspirin Not Indicated |
||||||||
|
Cardiovascular deaths║, MI, CVA
Cardiovascular deaths║
MI
Stroke¶ |
Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen Rofecoxib Naproxen |
3877 3878 3877 3878 3877 3878 3877 3878 |
23 15 6 5 12 4 8 7 |
2593 2596 2594 2597 2593 2597 2593 2997 |
0.89 0.58 0.23 0.19 0.46 0.15 0.31 0.27 |
0.65
0.83
0.33
0.87 |
(0.34, 1.25)
(0.25, 2.73)
(0.11, 1.03)
(0.32, 2.40) |
|
|
† Patient‑years at risk. ‡ Per 100 PYR. § Relative risk of naproxen with respect to rofecoxib from unstratified Cox model where the number of cases is at least 11, otherwise relative risk is ratio of rates. ║ Includes sudden death, unknown cause of death, fatal myocardial infarction, fatal stroke (hemorrhagic or ischemic), fatal subarachnoid hemorrhage, fatal primary intracranial hemorrhage, fatal gastrointestinal bleeding episode. ¶ Includes fatal and nonfatal ishemic strokes, and fatal or nonfatal hemorrhagic strokes. |
||||||||
136 These data showed that the relative risk of suffering a CVT serious adverse experience from taking naproxen rather than Vioxx was 0.42 (0.25, 0.72). This was represented in the text of the report as revealing that a person taking naproxen would have a 58% lower risk of encountering such an experience than a person taking Vioxx. Since the report sent to the FDA on 29 June 2000, there had been three further adjudications confirming myocardial infarctions in the Vioxx arm, taking the total to 20. The resulting relative risk figure (ie of taking naproxen rather than Vioxx) was shown in the report as 0.20 (0.07, 0.58). Within the aspirin‑indicated group, the risk of encountering any such experience was 0.20 (0.06, 0.71). It was not possible to calculate a relative risk figure for myocardial infarctions in that group, since there were no such events in the naproxen arm of the study. However, even amongst the patients where aspirin was not indicated, the relative risk of suffering a myocardial infarction from taking naproxen rather than Vioxx was very close to statistical significance: 0.33 (0.11, 1.03).
137 The applicant was critical of Merck for having expressed the VIGOR results as though the question were whether naproxen was cardioprotective. Approaching the matter from the perspective of Vioxx being the drug of interest and naproxen being the comparator, Prof Woodward recalculated the results as reported by Merck to the FDA. His summary table was the following:
Table 3.1 Relative risks from VIGOR, as reported by Merck to the FDA
|
|
No. of events |
|
Rates* |
|
Relative risk |
|
Outcome |
Rofe |
Naprox |
Rofe |
Naprox |
(95%CI) |
|
MI |
20 |
4 |
0.74 |
0.15 |
5.00 (1.72-14.29) |
|
All cardiac |
28 |
10 |
1.04 |
0.37 |
2.78 (1.35-5.88) |
|
Stroke1 |
11 |
9 |
0.41 |
0.33 |
1.22 (0.51-2.94) |
|
All CBV2 |
11 |
8 |
0.41 |
0.30 |
1.37 (0.56-3.45) |
|
CV death1 |
7 |
7 |
0.26 |
0.26 |
1.00 (0.35-2.86) |
|
APTC1 |
35 |
18 |
1.30 |
0.67 |
1.96 (1.10-3.45) |
|
Peripheral |
6 |
1 |
0.22 |
0.04 |
5.88 (0.73-∞) |
|
CVT2 |
45 |
19 |
1.67 |
0.70 |
2.38 (1.39-4.00) |
|
*per 100 person years |
|||||
|
CBV − cerebrovascular: CV= cardiovascular |
|||||
|
1 includes haemorrhagic stroke; 2 excludes haemorrhagic stroke |
|||||
The two entries which stand out in this table, and upon which the applicant placed most reliance, are the first and last – the risk of myocardial infarction and the risk of CVT events generally. In particular, given Merck’s emphasis upon the beneficial work of naproxen as the most likely explanation for the differences disclosed in VIGOR, it was put on behalf of the applicant that no interpretation of the results could conceivably justify a finding of a five‑fold increase in the risk of myocardial infarction without implicating Vioxx as a causative or contributory agent.
138 It seems that the three further myocardial infarctions to which I have referred in para 136 above had been known to Merck in July 2000. A memo from Dr Shapiro to Dr Reicin and others on 5 July 2000 referred to them (and to two other events, one in each arm). The authors of Bombardier (2000), however, including Drs Shapiro and Reicin, did not seek to amend their manuscript (which had been submitted in May). When the article was published in the New England Journal of Medicine in November 2000, it reported that “Myocardial infarctions were less common in the naproxen group than in the rofecoxib group (0.1 percent vs. 0.4 percent; 95 percent confidence interval for the difference, 0.1 to 0.6 percent; relative risk, 0.2; 95 percent confidence interval, 0.1 to 0.7).” The figures of 0.1% and 0.4% may be compared with the figures of 0.15% and 0.74% set out in the table in para 135 above. The latter did, but the former did not, contain the three additional myocardial infarctions.
139 In December 2005, the editors of the New England Journal published an “Expression of Concern”: Curfman (2005). They set out comparative statistics which showed that the true relative risk of suffering a myocardial infarction was 5.00 (1.68, 20.13), whereas the like figure if the three additional myocardial infarctions were omitted (ie as reported in Bombardier (2000), although not in those terms) was 4.25 (1.39, 17.37). They said:
The fact that these three myocardial infarctions were not included made certain calculations and conclusions in the article incorrect. … Lack of inclusion of the three events resulted in an understatement of the difference in risk of myocardial infarction between the rofecoxib and naproxen groups …. It also resulted in the misleading conclusion that there was a difference in the risk of myocardial infarction between the aspirin indicated and aspirin not indicated groups. In addition, the memorandum of July 5, 2000, contained other data on cardiovascular adverse events that we believe would have been relevant to the article. We determined from a computer diskette that some of these data were deleted from the VIGOR manuscript two days before it was initially submitted to the Journal on May 18, 2000. Taken together, these inaccuracies and deletions call into question the integrity of the data on adverse cardiovascular events in this article.
They said that they had asked the authors to submit a correction.
140 In response to those criticisms, both the “non-Merck” authors of Bombardier (2000) (of whom there were 11) and Drs Reicin and Shapiro wrote letters to the editor of the Journal, which were published in 2006. The former group expressed the view that their original article “followed appropriate clinical trial principles and does not require a correction.” They said that the non-inclusion of the three myocardial infarctions arose because 10 February 2000 had been specified in the cardiovascular data analysis plan as the cut-off date for reporting cardiovascular events. The correspondents said that the three additional myocardial infarctions “were neither in the locked database used in the analysis for the VIGOR paper nor known to us during the review process …” and added that “changing the analysis post hoc and after unblinding would not have been appropriate.” They pointed out, in effect, that omission or inclusion of the three myocardial infarctions would make no difference to the basic conclusion as to whether there was, or was not, a statistically significant relative risk of taking Vioxx rather than naproxen, whether in the aspirin-indicated group, in the non-aspirin-indicated group, or overall. They said:
Assessment of the relative risks and widely overlapping 95 percent confidence intervals does not suggest a difference in the conclusions that can be drawn from the original data and the updated data. We do not accept, therefore, that the addition of three myocardial infarctions leads to qualitatively different conclusions.
The correspondents also denied that the three myocardial infarctions had been deleted from the manuscript at any stage: they had never been there, because they were not part of the locked database.
141 In their letter, Drs Reicin and Shapiro made similar points to those referred to above. They defended the non-inclusion of the three myocardial infarctions on the basis that they had been reported after the cut-off date. They said:
[T]he editorial states that the omission of the post-cutoff date events rendered certain of the conclusions in the article incorrect. We disagree. The article clearly disclosed that there was a significant difference in the rates of myocardial infarction in the Vioxx and naproxen arms of the study and reported those rates as 0.4 and 0.1, respectively, with a relative risk reported as 0.2. The inclusion of the post-cutoff date myocardial infarctions changes the Vioxx rate to 0.5 but does not meaningfully change the relative risk or the conclusion that there was a significant difference between the two arms of the study.
At the general level, Drs Reicin and Shapiro said:
We believe it is important to emphasize that Merck consistently made full and appropriate disclosures of the cardiovascular results from VIGOR. First, Merck voluntarily announced the difference in thromboembolic event rates in March 2000. Second, Merck asked the FDA for accelerated review of labeling that would disclose the VIGOR. results. Third, the three post-cutoff myocardial infarctions were promptly disclosed to the FDA and were discussed at the public February 2001 FDA advisory committee. Finally, the additional data were included in subsequent communications about VIGOR, including journal articles, the FDA approved label, Merck press releases, and a transmittal letter to physicians that accompanied copies of the Journal VIGOR article provided by Merck.
I refer to what they described as the “FDA advisory committee” below.
142 In the same issue of the Journal, the editors referred to the correspondence they had received, but were unconvinced. They reaffirmed their expression of concern. They expressed the view that the article “did not accurately reflect the potential for serious cardiovascular toxicity with rofecoxib”.
The Merck pooled analyses
143 In her evidence, Dr Reicin said that MRL continued to analyse safety data arising from various Vioxx studies. Its most important source of data was that which came from the clinical trials of Vioxx. During 2000, Dr Reicin and her colleagues undertook a pooled analysis of cardiovascular clinical trial data from the Phase IIb to Phase V clinical trials of Vioxx that were of at least 4 weeks’ duration and in which placebo and/or non-selective NSAIDs were the comparators. That was the subject of a statistical data analysis plan prepared on 12 October 2000. In the introduction to that plan, it was said that a review of the clinical trials databases for Vioxx in patients with osteoarthritis and early Alzheimer’s disease demonstrated that therapy with rofecoxib was accompanied by rates of cardiovascular events that were comparable to those arising under the administration of placebo or non-selective NSAIDs. It was said that those studies differed from the VIGOR study in three respects: they evaluated different patient populations; in most cases, they compared rofecoxib either to placebo or to non-selective NSAIDs with weaker antiplatelet properties than naproxen; and, in some cases, they allowed low‑dose aspirin therapy in patients at risk for thrombotic cardiovascular events. It was, therefore, desired to carry out meta‑analyses on all ongoing Merck coxib trials (including also another drug of Merck’s, etoricoxib) to maximize the amount of information available to assess the comparative effects of non‑selective NSAIDs, coxibs, and placebo on CVT events. It was said that the primary objective of the analysis was to estimate the incidence rates of CVT events in patients treated with coxibs in comparison to those treated with non-selective NSAIDs and, separately, in comparison to those treated with placebo. There were to be no specific hypotheses because the objectives concerned safety and a “consensus on definition of comparability bounds for clinically important safety events such as thrombotic cardiovascular events would be difficult to establish”, and because the analysis was “exploratory due to the inconsistency of the VIGOR results compared with those of the other rofecoxib studies”.
144 The primary endpoint of the analysis was to be the Antiplatelet Trialists’ Collaboration (APTC) definition of thrombotic cardiovascular events. That definition included cardiovascular death, myocardial infarction, stroke (both ischaemic & haemorrhagic), and death due to unknown cause or due to bleeding. However, the plan proposed:
It is recognized that inclusion of bleeds in the primary endpoint could reduce assay sensitivity to detect negative effects of COXIBs since VIOXX has been shown to reduce [gastrointestinal] bleeds. However, given that the primary endpoint reflects a traditional definition of a set of relevant events by a consensus of experts, it was chosen as primary for this analysis. Recognizing the potential for reduction in assay sensitivity of the primary endpoint, a secondary endpoint which excludes the bleed events and hemorrhagic strokes will be provided to remove the potential for such reduction.
The secondary endpoint was based on confirmed CVT events in accordance with the CV SOP. They were identified as:
· Confirmed thrombotic cardiovascular serious adverse experiences (secondary endpoint; terms defined in CV Adjudication SOP, Section K, with the exception of hemorrhagic cerebrovascular stroke or hemorrhagic change, fatal or non‑fatal, and non-thromboembolic event)
· All confirmed cardiac events
· Confirmed myocardial infarction
· All confirmed cerebrovascular events (excluding hemorrhagic stroke)
· All confirmed peripheral vascular events
· Confirmed cardiovascular deaths [ie including confirmed adjudicated cardiovascular deaths, confirmed sudden or unexplained death, or death associated with bleeding events]
· Confirmed stroke (ischaemic and hemorrhagic)
It was said:
The typical approach to evaluating a large set of related endpoints is to create a hierarchy by designating primary, secondary, and exploratory sets of endpoints. This is the approach taken here. The interpretation of results in this scenario is to focus on the primary endpoint’s results and use the results of secondary and exploratory endpoints as corroborative evidence to support the overall conclusion regarding the primary endpoint. This is intended to address issues of multiplicity of endpoints, where false positive observations increase with increased numbers of endpoints. The situation, however, in evaluating safety endpoints is somewhat different in that the assessment of safety is a screening exercise. Hence, a level of importance must be given to the integrated clinical assessment of results of all endpoints. This interpretation must be made with caution in consideration of the multiplicity of endpoints issue. Furthermore, in the circumstance of this DAP, the primary endpoint is a composite which could mask effect(s) in individual components. Thus, the analyses of the individual components identified above will be given appropriate clinical attention together with consideration of consistency of results across study blocks and across related endpoints in the formulation of appropriate conclusions.
145 Merck provided pooled analyses of the cardiovascular results of its Vioxx trials to the FDA on four occasions between June 2000 and September 2004: on 8 January 2001, on 12 July 2001, on 22 May 2002 and on 22 March 2004. The studies covered by those analyses fell into three broad groups. The first group consisted of Phase IIb and III studies of patients with rheumatoid arthritis, including, in the third and fourth analyses, VIGOR. The second group consisted of Phase IIb (one study) and III studies of patients with osteoarthritis. It was a characteristic of these arthritis studies that the comparator was usually a non‑selective NSAID. Although a number of the studies had placebo arms, since the patients concerned would suffer from pain and inflammation in the absence of medication of some kind, exposure to placebo was generally limited in time. The third group consisted of Phase III studies of patients with, or at risk of, Alzheimer’s disease, together with a number of other relatively small studies of patients with low back pain and, for some of the analyses, prostatitis and familiar adenopolypsis. In this third group, it was only the Alzheimer’s studies that were the subject of any critical attention by the applicant in this proceeding. They were important in the pooled analyses generally because they provided the bulk of the placebo‑controlled data.
146 There were three Alzheimer’s disease studies. Protocol 078, which commenced on 29 April 1998 and ran until 23 April 2003, was a placebo‑controlled, parallel‑group, double‑blind study to evaluate the effect of Vioxx 25 mg per day on the prevention of Alzheimer’s disease and cognitive decline in patients of 65 years or older with mild cognitive impairment. Patients were randomised to receive Vioxx 25 mg or placebo for four years or until 220 cases of clinically‑diagnosed probable or possible Alzheimer’s disease were observed, whichever occurred later. Over the course of the study, 725 patients were enrolled in the Vioxx arm, and 732 patients were enrolled in the placebo arm. 117 and 148 patients in these arms, respectively, completed the study. Protocol 091, which commenced on 10 February 1999 ran until 30 November 2000, was a placebo‑controlled, parallel‑group, 15‑month, double‑blind study to evaluate the efficacy and safety of Vioxx 25 mg to slow the progression of symptoms of Alzheimer’s disease. Patients aged 50 years or older were randomised to receive Vioxx 25 mg or placebo daily for 12 months, followed by an additional 3‑month treatment phase in which 90% of the patients initially assigned to Vioxx would be treated with placebo, while the other patients would remain on their initial treatment. Over the course of the study, 346 patients were enrolled in each of the Vioxx and placebo arms. 253 and 268 patients in these arms, respectively, were continuing at the 12‑month point. Protocol 126, which commenced on 3 March 2000 and which was to have run until 31 May 2001, was of identical design to Protocol 091. Over the course of the study, 382 patients were enrolled in the Vioxx arm, and 376 patients were enrolled in the placebo arm. The study was terminated prematurely on 31 March 2001 because the results of Protocol 091 failed to demonstrate the efficacy of Vioxx in slowing the progression of Alzheimer’s disease.
The first pooled analysis
147 The first pooled analysis was submitted to the FDA on 8 January 2001 as an amendment to the Vioxx Investigational New Drug Application. It was described as an “Interim Cardiovascular Meta‑Analysis”. It used the APTC endpoints only. The studies included in the analysis were identified as follows:
The data for this analysis derive from all rofecoxib Phase IIb through V trials conducted by Merck & Company that were of at least 4 weeks duration, excluding those performed in healthy volunteers, and where there were comparative data between rofecoxib and either NSAIDs and/or placebo. Trials that compared rofecoxib to celecoxib, another selective COX-2 inhibitor, were excluded. Data from treatment periods using doses of rofecoxib <12.5 mg (sub‑therapeutic doses) were excluded from analysis; doses ≥12.5 mg were combined for analysis. The studies included in this initial meta‑analysis were all trials completed and unblinded in early September, 2000, as well as 2 ongoing trials in Alzheimer’s patients [Protocols 078 and 091]. Interim data from the 2 Alzheimer’s studies were included using a cutoff of September 15, 2000.
It may be noted that Protocol 126 was not part of this analysis.
148 The analyses were conducted according to Merck’s “modified intention‑to‑treat principle”. An event which occurred up to the fourteenth day after the discontinuation of treatment by the patient concerned was counted in the analysis. In each instance, only the first eligible event for an individual patient was counted. As to the inclusion of adjudication or investigator‑reported events, it was stated:
Adjudicated data were used wherever possible. Protocols 068 [rheumatoid arthritis] and 069 [osteoarthritis] were completed prior to the institution of the Cardiovascular Adjudication SOP in the rofecoxib program, and adjudications were not complete for Protocols 096, 097 [both rheumatoid arthritis] and the ongoing Alzheimer’s trials at the time of this analysis. For these studies, all reported events of CV death, MI, and stroke qualified for analysis. In the other studies, only confirmed adjudicated events qualified for CV death, MI, and stroke. In all studies, all deaths due to unknown cause or due to bleeding qualified for analysis. For studies which had not undergone or completed adjudication, all deaths were reviewed by a Merck physician in a blinded fashion to determine whether or not an APTC endpoint had occurred.
149 The data in the pooled analyses were presented in a number of ways in the report of 8 January 2001. The tabular presentation that will suffice for present purposes was the following:
|
Indication for Treatment |
Rofecoxib |
Comparator |
Relative risk (95% CI)3 |
||
|
N |
Cases/PYR1 (Rate2) |
N |
Cases/PYR1 (Rate2) |
||
|
Rofecoxib vs naproxen |
|||||
|
RA OA Alz/LBP Total |
6057 3026 0 9083 |
46/3947 (1.17) 11/675 (1.63) – 57/4622 (1.23) |
4859 3011 0 7870 |
20/3078 (0.65) 7/665 (1.05) – 27/3742 (0.72) |
1.74 (1.02, 2.96) 1.55 (0.60, 4.00) – 1.69 (1.07, 2.69) |
|
Rofecoxib vs other [ie non-naproxen] nonselective NSAIDs |
|||||
|
RA OA Alz/LBP Total |
0 4549 0 4549 |
– 21/1934 (1.09) – 21/1934 (1.09) |
0 2755 0 2755 |
– 14/984 (1.42) – 14/984 (1.42) |
– 0.79 (0.40, 1.55) – 0.79 (0.40, 1.55) |
|
Rofecoxib vs placebo |
|||||
|
RA OA Alz/LBP Total |
1622 3165 1503 6290 |
3/337 (0.89) 12/655 (1.83) 18/1197 (1.50) 33/2189 (1.51) |
989 1215 1278 3482 |
1/201 (0.50) 3/232 (1.30) 28/1246 (2.25) 32/1678 (1.91) |
1.78 (0.14, 93.70) 1.53 (0.43, 5.44) 0.68 (0.37, 1.23) 0.84 (0.51, 1.38) |
|
Rofecoxib vs other [ie non-naproxen] nonselective NSAIDs/placebo |
|||||
|
RA OA Alz/LBP Total |
1622 4550 1503 7675 |
3/337 (0.89) 21/1938 (1.08) 18/1197 (1.50) 42/3472 (1.21) |
989 3750 1278 6017 |
1/201 (0.50) 17/1201 (1.42) 28/1246 (2.25) 46/2648 (1.74) |
1.78 (0.14, 93.70) 0.81 (0.42, 1.53) 0.68 (0.37, 1.23) 0.76 (0.50, 1.16) |
|
Rofecoxib vs any nonselective NSAIDs/placebo |
|||||
|
RA OA Alz/LBP Total |
6057 7576 1503 15136 |
46/3947 (1.17) 32/2613 (1.22) 18/1197 (1.50) 96/7758 (1.24) |
5174 6761 1278 13213 |
21/3204 (0.66) 24/1866 (1.29) 28/1246 (2.25) 73/6316 (1.16) |
1.72 (1.02, 2.90) 1.00 (0.59, 1.71) 0.68 (0.37, 1.23) 1.10 (0.81, 1.50)4 |
|
1 Patient-years at risk 2 Per 100 PYR 3 Relative risk of rofecoxib with respect to comparator from Cox model stratified by indication for totals and unstratified elsewhere when number of cases is at least 11, otherwise relative risk is ratio of rates. 4 This estimate is provided for information only since it represents pooling of heterogenous cohorts. |
|||||
The respondents stressed the figures shown in bold in the first three panels in the right hand column of this table. They submitted (as the figures indicate) that only where the comparator was naproxen was it evident that Vioxx was significantly riskier than the comparator: indeed, where the comparator was another NSAID or placebo, the non‑significant result tended to indicate that Vioxx was the less risky. In her evidence, Dr Reicin said that this interim data did not show an increased risk on Vioxx compared to placebo or to NSAIDs other than naproxen. She believed that the results were similar to what had been seen when she originally examined the VIGOR, the Phase IIb/III osteoarthritis and the Alzheimer’s data.
150 The report of 8 January 2001 claimed that the analyses referred to justified the following “critical findings”:
1. A meta‑analysis of all placebo‑controlled trials of rofecoxib demonstrated that the therapy with rofecoxib and placebo resulted in a comparable incidence of the APTC endpoint. Thus, there was no evidence to suggest that therapy with rofecoxib is associated with an increased risk of thrombotic cardiovascular events. These results are of particular interest because the Alzheimer studies which contributed the majority of patient years and events to this analysis is an elderly cohort (mean age 75) with the highest baseline incidence of traditional risk factors in the rofecoxib clinical program. This elderly cohort serves as an excellent patient population in which to affirm the cardiovascular safety of rofecoxib.
2. A meta‑analysis of all trials comparing rofecoxib with (non‑naproxen) non‑selective NSAIDs that do not demonstrate potent and sustained antiplatelet effects (diclofenac, ibuprofen, nabumetone), revealed that therapy with rofecoxib or these compounds was associated with comparable incidences of the combined APTC endpoint. These results indicate that therapy with rofecoxib is not accompanied by an increased risk of thrombotic cardiovascular events compared with non‑selective NSAIDs which lack potent and sustained anti‑platelet effects.
3. An analysis of heterogeneity suggested that the cardiovascular findings of the meta‑analysis of naproxen‑controlled rofecoxib trials and the meta‑analysis of trials comparing rofecoxib to non‑selective NSAIDs other than naproxen may have been different. Specifically, the relative risk of 1.69 seen in the comparison of rofecoxib to naproxen tended to be different than the relative risk of 0.79 seen in the comparison of rofecoxib to non‑selective NSAIDs other than naproxen. These results support the contention that the imbalanced rates of thrombotic cardiovascular events observed in the clinical trials that compared rofecoxib with naproxen may have been due to a reduced rate of events in the naproxen group rather than an increased rate in the rofecoxib group. A potential mechanistic explanation for this is naproxen’s ability to provide sustained nearly complete inhibition of platelet function and thus serve as a cardioprotective agent, rather than an adverse effect of rofecoxib. Clinical pharmacology studies discussed in the original rofecoxib NDA and in the VIGOR cardiovascular report demonstrated that naproxen, but not diclofenac, nabumetone, or ibuprofen produces near‑complete, sustained antiplatelet effects through its potent inhibition of COX-1 throughout the dosing interval, effects that are similar to those observed with aspirin, an established cardioprotective agent. Rofecoxib, conversely, in keeping with its COX-2 selectivity, does not influence platelet function and thus would not be expected to be protective in patients at risk for cardiovascular adverse events.
The Arthritis Advisory Committee meeting
151 There was to be a meeting of the FDA Arthritis Advisory Committee on 8 February 2001. The report of 8 January 2001 would be considered at that meeting. Also for the purpose of that meeting, on 4 January 2001 MRL sent to the FDA a background paper which recognised the “theoretical possibility that selective COX-2 inhibitors, because they inhibit prostacyclin production but not platelet function, might result in proaggregatory effects”. However, the paper expressed the following conclusions:
– The rates of thrombotic cardiovascular serious adverse experiences in patients taking rofecoxib are similar to those in patients taking the nonselective NSAIDs ibuprofen, diclofenac, or nabumetone.
– The rates of thrombotic cardiovascular serious adverse experiences in patients taking rofecoxib are similar to those in patients taking placebo, including elderly patients at relatively high risk for thrombotic cardiovascular serious adverse experiences.
– The rates of thrombotic cardiovascular serious adverse experiences are lower in patients treated with naproxen compared to rofecoxib. This difference in rates does not appear to be due to a prothrombotic effect of rofecoxib but rather a cardioprotective benefit of naproxen due to its potent and sustained antiplatelet effects.
152 In the period leading up to the FDA Arthritis Advisory Committee meeting, Dr Scolnick was clearly worried that the issue of the proper interpretation of the VIGOR results remained unresolved. On 31 January 2001, he sent an email which contained the following passage to the Chief Executive Officer of Merck, Mr Raymond Gilmartin, and another senior executive:
On Monday I will show you the essence, an update, of the data that supports Vioxx is safe in the [cardiovascular] sense. But I want to point out to all of you at one time that … there is no way to prove that in patients with rheumatoid arthritis that ALL of the difference between Vioxx and naproxen is due to the benefit of naproxen. IT IS IMPOSSIBLE TO PROVE THIS; IT IS IMPOSSIBLE TO KNOW THIS WITH CERTAINTY. It is likely if not certain that our label will state the data from Vigor. It is even likely that words will be used to say that it is not clear if the effect is purely due to a protective effect of naproxen in this [rheumatoid arthritis] patient population. When the study results came out last year, this fact was patently clear. Since then we have reduced the uncertainty to this very salient point. But it is impossible to dismiss the point. The FDA will NEVER allow it to be fully dismissed. There will be great adverse publicity at the meeting. Sidney Wolfe now is on the agenda after both us and the FDA on our day. I am trying to find out whether he has asked for time on their day also. In any case we need to face the reality of the situation and manage it.
As far as I can infer from the evidence “our day” was the day set aside by the committee for the consideration of the Vioxx data, and “their day” was the corresponding day on which Celebrex would be dealt with.
153 On 1 February 2001 (and, I would infer, for the purposes of the Advisory Committee meeting on 8 February 2001), Dr Shari Targum, a medical officer within the division of cardio‑renal drug products in the FDA, prepared a memorandum for the division of anti‑inflammatory drug products. It contained a review of the cardiovascular safety database for Vioxx. Dr Targum’s review dealt at length with the VIGOR trial, and with two other studies, Protocols 085 and 090, each a placebo‑controlled RCT to evaluate the safety and efficacy of Vioxx 12.5 mg in patients with osteoarthritis of the knee, where nabumetone 1000 mg was the comparator. Of the VIGOR results, Dr Targum said:
The cardiovascular thrombotic event rates, while not high, were significantly different between the two groups; most striking were the myocardial infarction event rates. Thus, to this Medical Reviewer, there are more cardiovascular thrombotic events in the rofecoxib group than in the naproxen group; the time‑to‑event curves are different, favoring naproxen. This Medical Reviewer is concluding that there is an increased risk of cardiovascular thrombotic events, particularly myocardial infarction, in the rofecoxib group compared with the naproxen group. More difficult is the question of a safety signal for rofecoxib. As there is no placebo group, it will be difficult to assess the CV thrombotic risk with rofecoxib use compared with no therapy at all.
Dr Targum noted that Merck’s claim that the difference in myocardial infarctions as between the two arms of VIGOR was primarily due to the antiplatelet effect of naproxen was “not supported by any prospective placebo‑controlled trials with naproxen”. Of Merck’s claim that the majority of CVT events in the VIGOR trial occurred in patients who should have been taking aspirin for cardioprotection, Dr Targum said that the data were consistent in the sense that, even in patients who were not aspirin‑indicated, there were increased events in the Vioxx arm. As to Merck’s claim that patients with rheumatoid arthritis were particularly at risk for CVT events, Dr Targum noted that there was some literature which dealt with that aspect, but said that, even if there were substance in the claim, “one is still faced with the difference in cardiovascular events between rofecoxib and naproxen”. As to the labelling that would properly address cardiovascular risks, Dr Targum said that it would be difficult to imagine the inclusion of the VIGOR results in the labelling of Vioxx “without mentioning cardiovascular safety results in the study description as well as the Warnings sections”.
154 In evidence is a transcript of the deliberations of the FDA Arthritis Advisory Committee meeting of 8 February 2001. Apart from the members of the committee as such and scientific staff of the FDA, present were two consultants to the committee, two members of the Cardiovascular and Renal Drugs Advisory Committee, a member of the Gastrointestinal Drugs Advisory Committee, a member of the Endocrinologic and Metabolic Drugs Advisory Committee, a biostatistician, a guest expert, a consumer representative and representatives of Merck, including Dr Reicin. The first section of the meeting was, it seems, held in closed session, and the second section was open to the public. The main concern of the discussion was to settle the changes, if any, that should be required on the labelling of Vioxx (in the United States) in light of the VIGOR results.
155 In her presentation to the committee, Dr Reicin acknowledged that the inhibition of the production of prostacyclin without at the same time blocking the activity of platelets may have clinical significance. But she expressed the conclusion that the totality of the data then available to MRL “demonstrated that the risk of sustaining a cardiovascular event on rofecoxib is similar to placebo and to NSAIDs without sustained anti‑platelet activity [and that the] ... reduction in events on naproxen compared with rofecoxib [appeared] to be the outlier.”
156 It is clear from the transcript of the meeting of the committee that the potential for Vioxx to be prothrombotic was recognised as a real one, but the consensus seemed also to be that the antiplatelet activity of naproxen was to an extent responsible for the results of VIGOR. As I read the transcript, there was also a general acceptance that the VIGOR trial had established the gastrointestinal superiority of Vioxx over naproxen. There was a view that more work needed to be done on the cardiovascular side. Towards the end of the meeting, a member of the Cardiovascular and Renal Drugs Advisory Committee, Dr Steven Nissen, expressed a view which attracted general support from those present, and was the basis of the committee’s ruling as to the label changes required to be made in the United States for Vioxx. Dr Nissen said:
Briefly, I think what I would say in the label is that there was an excess of cardiovascular events in comparison to naproxen, that it remains uncertain whether this was due to beneficial cardioprotective effects of naproxen or prothrombotic effects of the agent, and leave it at that, that basically we don’t know the reason. We do know there was a difference. That awareness should be made available to the prescriber and to the consumer, but without necessarily a final judgment as to the reasons for that difference.
157 According to Dr Reicin, the FDA and Merck worked together to develop revised labelling for Vioxx. Dr Reicin said that the FDA approved revised wording for the United States package insert in April 2002. At the same time, the FDA approved Vioxx for a new indication, the relief of the signs and symptoms of rheumatoid arthritis in adults. The FDA approved Vioxx for the acute treatment of migraine attacks in adults in March 2004 and for the relief of the signs and symptoms of juvenile rheumatoid arthritis in August 2004.
The Konstam article
158 The data submitted by Merck to the FDA on 8 January 2001 were the subject of an article prepared, largely within MRL, for submission to a peer‑reviewed journal. It seems that the article was drafted in the weeks leading up to August 2001. The article was ultimately published as Konstam (2001). The authors were Marvin A Konstam MD, Matthew R Weir MD, Alise Reicin MD, Deborah Shapiro DrPh, Rhoda S Sperling MD, Eliav Barr MD and Barry J Gertz MD PhD. All except the first two were employees of Merck, and worked at MRL. In accordance with what appears to have been practice within MRL, drafts of the article were circulated within that organisation for comment. One of those so consulted was Dr Morrison. He appears to have been sent a fairly early draft by Dr Sperling on 6 July 2001 (more of a “shell” as she described it). Although counsel for the applicant supposed that the draft might have been of the article ultimately published as Weir (2003), I would infer, from the timing, that the draft was of what became Konstam (2001). Dr Morrison’s comments, sent to Dr Sperling on 8 July 2001, included the following:
My preference is to do the biochemical stuff first as a basis for theoretic risk of COXIB vs placebo – that ([to] me) is the real issue for physicians. Then do all placebo controlled clinical data and nail really, really hard that there is no prothrombotic effect – Vioxx does not cause [myocardial infarction]s. To me, that is the key message we need to emphasize. … [Cardiovascular] thrombotic events seen in VIGOR consistent with a lack of effect on COX-1 mediated thromboxane biosynthesis, the major determinant of platelet aggregation.
There seems no reason to think that this was not how Dr Morrison actually viewed the science at the time.
159 What appears to have been a fairly final draft of the article was circulated for comment on 15 August 2001. Dr Morrison provided his comments by email dated 17 August 2001. By reference to a table which contained the data set out in the table in para 149 above, Dr Morrison wondered whether, in the placebo comparison, the Alzheimer’s data might be “the outlier”. Looking at the arthritis data, he noted that the relative risk (point estimate) figure for both the osteoarthritis and the rheumatoid arthritis comparisons were strikingly consistent as between the placebo comparison and the naproxen comparison. In this respect he wondered whether the low figure for the osteoarthritis non‑naproxen NSAID comparison might again be “the outlier”. He questioned whether the “LBP” (low back pain) data should be grouped with those from the Alzheimer’s trials. He also questioned the pooling of the arthritis data with the Alzheimer’s data, noting that the draft article had in this respect contented itself with working by reference to the absence of statistical heterogeneity, without considering the “biologic differences between patient populations”.
160 Summarising, Dr Morrison said:
I guess what I am saying is that the data appears to have been interpreted to support a preconceived hypothesis rather than critically reviewing the data to generate hypotheses. The Second line of the Discussion says “There was no evidence (emphasis mine) that rofecoxib was associated with excess CV events compared with either placebo or non‑naproxen NSAIDs” – that seems wishful thinking, not a critical interpretation of the data. Of course if you look at Figure 1, the 95% Cls for all comparisons include the possibility that there is a real increases [sic] risk on rofecoxib.
The “figure 1” to which Dr Morrison referred was the following:
In this representation of the relative risk, the lower point of the white triangle is the point estimate, the size of the triangle is an approximate indication of the amount of data which lies behind the risk figure, and the horizontal black line is an indication of the 95% confidence interval.
161 A senior statistician in MRL, Dr Jim Bolognese, replied by email the same day. He accepted that Dr Morrison had made some good points, but said that his suggestions, while plausible, were “based on too‑small numbers”. He demonstrated that, if there were only one more event observed in the placebo arms for each of the osteoarthritis and rheumatoid arthritis groups, the relative risks for them would be in line with the corresponding figures in the Alzheimer’s and non‑naproxen NSAID data. He continued:
So, I think the critical review of the data is largely based on the bulk of the data which divide into two independent large chunks – rofe vs. placebo in ALZ, and rofe vs. non‑naproxen in OA, both of which consistently suggest no negative impact of rofe, So, I think the conclusion is appropriate.
Dr Bolognese’s response was supported by Dr Shapiro. In a final email also sent on 17 August 2001, Dr Morrison made it clear that he was not entirely satisfied with the statisticians’ explanations, but added that his comments were “discretionary” and that they were submitted “only as a surrogate for a minimally informed reviewer”.
162 In the article as published in November 2001 (Konstam 2001), the authors said that “there was no evidence that rofecoxib was associated with excess CV events compared with either placebo or non‑naproxen NSAIDs”. They continued:
Most importantly, the results of the rofecoxib relative to placebo comparison demonstrated a comparable risk of APTC events in both groups. Because placebo was given for a limited duration in the osteoarthritis, rheumatoid arthritis, and chronic low back pain studies, the majority of patient‑years came from the Alzheimer’s trials, whose populations included predominantly elderly, male patients. These data, which were obtained from studies directly comparing rofecoxib and placebo, are most reassuring that there is no evidence for any increased risk of CV thrombotic events with rofecoxib. The results of the rofecoxib relative to non‑naproxen NSAID comparison also demonstrated comparable risks of APTC events in both groups. However, non‑naproxen NSAIDs were only studied in osteoarthritis patients, and there were insufficient data to differentiate the effects of the individual NSAIDs studied.
The second pooled analysis
163 The second pooled analysis was provided in the body of a Safety Update Report provided by Merck to the FDA on 12 July 2001 in response to a request from the FDA made on 6 April 2001. The studies included in the analysis were identified as follows:
All studies that were 4 weeks or more in duration, that included a placebo or non‑naproxen NSAID comparator, and had a complete and frozen database by 16‑Mar‑2001 were included. The exception to this are the ongoing (Protocol 078) or recently ended (Protocol 126) Alzheimer’s Disease Studies for which thrombotic cardiovascular serious adverse experiences reported to Merck & Co., Inc. by 16‑Mar‑2001 were included in the Investigator Reported Cardiovascular Event analyses. Data adjudicated by 15‑May‑2001 from Protocols 078, 091, and 126 were included in the APTC and Confirmed Cardiovascular Event analyses. A new study in patients with prostatitis (Protocol 118) is included in this update but was not included in the prior meta‑analysis.
164 There were no data, additional to that provided to the FDA on 8 January 2001, based on a comparison of Vioxx with naproxen. The data based on a comparison of Vioxx with other NSAIDs were derived only from the osteoarthritis studies, and showed an overall relative risk (of taking Vioxx rather than an NSAID) of 0.84 (0.45, 1.63). The data based on a comparison of Vioxx with placebo were presented in tabular form and, showing only the overall results from each group, were as follows:
|
Indication for Treatment |
Study Group |
Rofecoxib |
Placebo |
Relative Risk (95% CI)▫ |
||
|
N |
Cases/PYR† (Rate‡) |
N |
Cases/PYR† (Rate‡) |
|||
|
Rofecoxib vs Placebo |
||||||
|
RA |
All |
1622 |
3/337 (0.89) |
989 |
1/201 (0.50) |
1.78 (0.14, 93.70) |
|
OA |
All |
3165 |
12/655 (1.83) |
1215 |
3/232 (1.30) |
1.42 (0.38, 7.81) |
|
Alz/LBP/ Prostatisis |
All |
2012 |
25/1526 (1.51) |
1737 |
30/1667 (1.80) |
0.84 (0.46, 1.49) |
|
Total |
All |
6799 |
38/2518 (1.51) |
3941 |
34/2099 (1.62) |
0.93 (0.57, 1.53) |
|
† Patient years at risk. ‡ Per 100 PYR. ▫ Relative risk of rofecoxib with respect to comparator is calculated from ratio of rates. |
||||||
The respondents relied on these data as indicating an absence of any statistically significant evident risk of taking Vioxx by comparison with placebo or NSAIDs other than naproxen.
165 The report did not present a pooled analysis of CVT events as such, justifying the use of the APTC endpoint as follows:
The choice of the APTC endpoint was prespecified for this analysis for the following reasons: (1) as noted above, it is the most commonly accepted endpoint used in trials evaluating antithrombotic agents, and this was the endpoint suggested by a group of consultants after reviewing the initial VIGOR results; and (2) investigator‑reported events were included from studies and patients that will not or have not yet completed the adjudication process. Since cardiovascular and unknown cause of death, myocardial infarction, and cerebrovascular accident are “hard endpoints”; the correlation is quite high between investigator‑reported events and events confirmed by the Adjudication committee. Therefore, use of the APTC endpoint helped ensure consistency between trials that used adjudicated data and those which used investigator reported data.
The report recognised that the inclusion of some haemorrhagic events in the APTC endpoint might have the effect of compromising the ability of the analysis to reveal any prothrombotic tendencies of Vioxx, and dealt with the problem in these terms:
Hemorrhagic strokes are an APTC endpoint that may not reflect an underlying thrombotic event. Inclusion of bleeding events in the primary endpoint could reduce the sensitivity to detect a prothrombotic effect of COX-2 selective inhibitors. However, given that the primary endpoint reflects a traditional definition of a set of relevant events by a consensus of experts, it was chosen as primary for this analysis. Since the incidence of hemorrhagic strokes was quite low (3 hemorrhagic strokes on rofecoxib and 2 on placebo), exclusion of these events would not change the conclusions of the primary analysis.
166 Notwithstanding that rider, the report did deal with some aspects of the CVT endpoint. Data from the Alzheimer’s studies (where the comparator was placebo) were set out in two tables, the first showing the confirmed events, and the second showing the investigator‑reported events. I have combined those tables to show the relative risk data only in the following form:
|
Indication for Treatment |
Study Group |
Relative Risk (5% CI)* Confirmed |
Relative Risk (95% CI)* Investigator-Reported |
|
Rofecoxib vs Placebo |
|||
|
Alzheimer’s |
Protocol 078 Protocol 091 Protocol 126 |
0.82 (0.41, 1.59) 0.41 (0.10, 1.34) 1.03 (0.19, 5.51) |
0.83 (0.51, 1.35) 0.98 (0.42, 2.25) 1.65 (0.47, 6.40) |
|
Total |
All |
0.72 (0.42, 1.21) |
0.93 (0.63, 1.37) |
|
* Relative risk of rofecoxib with respect to comparator is calculated from ratio of rates. |
|||
167 The report described the “principal findings” of the analysis referred to in it as follows:
First, a meta‑analysis of all placebo‑controlled trials with rofecoxib demonstrated comparable cardiovascular event rates for rofecoxib and placebo, whether these events were defined by the investigator or by the adjudication process. This observation is of special interest in that it includes a significant proportion of elderly patients at increased risk for cardiovascular disease. Furthermore, a meta‑analysis comparing rofecoxib with non‑naproxen NSAIDs also demonstrated comparability in the frequency of cardiovascular event rates. These results indicate that rofecoxib treatment is not associated with an increase in the risk of cardiovascular events across a broad population of patients compared with the NSAIDs that do not demonstrate potent and sustained anti‑platelet effects. Finally, the results of this meta‑analysis are consistent with those observed in the interim analysis submitted Jan-2001.
The report summarised the data with respect to CVT events in the following terms:
The Adjudication committees also evaluated thrombotic cardiovascular serious adverse experiences that did not meet the strict APTC criteria. The Adjudication committees confirmed that in the Alzheimer’s Disease studies there were 25 rofecoxib patients with thrombotic cardiovascular serious adverse experiences during 1461 patient‑years for a rate of 1.71 confirmed events per 100 patient‑years. This is similar to the rate of 2.39 events per 100 patient‑years with placebo (39 events/1634 patient‑years). The relative risk is 0.72 with a 95% CI of (0.42, 1.21). In each of these 3 large studies, the rate for rofecoxib was statistically indistinguishable from that with placebo. There was no apparent difference when cardiac events or cerebrovascular events were examined separately.
Broadening the analysis to include potential thrombotic cardiovascular events reported by the investigators but not confirmed by adjudication does not change the overall result. In Protocols 078, 091, and 126, investigators reported 52 rofecoxib patients with thrombotic cardiovascular serious adverse experiences during 1450 patient‑years for a rate of 3.59 events per 100 patient‑years. This is similar to the rate of 3.84 events per 100 patient‑years with placebo (62 events/1615 patient‑years). In each of these 3 large studies, the rate for rofecoxib was similar to that with placebo. There was no apparent difference when cardiac events or cerebrovascular events were examined separately.
The conclusion expressed in the report was that Vioxx had a favourable safety profile in the osteoarthritis population, including patients aged 80 years and older. There were said to be “no findings on new clinically important adverse experiences with the longer durations of exposures”. It was said that Vioxx had a cardiovascular safety profile comparable to that of placebo, and that the risk of sustaining a thrombotic cardiovascular event was similar in patients treated with Vioxx, placebo, or an NSAID (other than naproxen) “that [lacked] potent and sustained inhibition of platelet function”.
The FDA warning letter
168 On 17 September 2001, the Public Health Service of the FDA wrote a “warning letter” to Merck with respect to promotional activities and materials for the marketing of Vioxx in the United States. That particular aspect of the letter is not relevant to the allegations in this proceeding, but the applicant relied on the letter as part of the material which was said to have put Merck on notice of the cardiovascular risks associated with the consumption of Vioxx, and of the absence of any scientific support for the naproxen theory advanced by way of explanation of the results of the VIGOR trial. If the terms of the letter are to be believed, Merck had procured the production of audio material for promotional conferences in which it was said that naproxen was a “wonderful platelet inhibitor”. It was said that naproxen “inhibits platelets identically to aspirin” and that, while the binding of naproxen was reversible, “the effect on platelets and bleeding time is identical in terms of its effect and therefore functions as a wonderful drug for cardiovascular prophylaxis”. The FDA letter referred to a number of other marketing representations which had, it was alleged, been made by Merck and which conspicuously overreached the then known science.
169 The terms of the FDA letter were put to Dr Reicin under cross‑examination. She said that the letter had come to her attention after her return from leave in October 2001, and that she and others had visited the offices of the FDA to brief them about MRL’s position with respect to the VIGOR results, and to Vioxx generally. Dr Reicin said that the people responsible for the FDA letter were not scientists. The matter was resolved by Merck agreeing to distribute a letter of correction to all those who had been present at the conferences where the offending material had been used. That letter was sent in November 2001 and, relevantly to the matter of cardiovascular risk, it stated:
Alternative interpretations have been proposed for the difference in the rates of myocardial infarctions … in the Vioxx treatment group in comparison with the naproxen treatment group. Possible explanations include that Vioxx increased the myocardial infarction rate or naproxen decreased the myocardial infarction rate. The underlying reason for the difference has not been established in prospectively designed clinical studies.
The Reicin article
170 A more limited analysis than Konstam (2001) was that submitted for publication on 2 July 2001 and published in January 2002 as Reicin (2002). The authors looked at the data from Merck’s Phase IIb/III osteoarthritis safety database, which included all patients participating in 8 double‑blind, placebo, and active‑comparator controlled studies of osteoarthritis conducted from 1995 to 1998. The events of interest were investigator‑reported, and included –
… those that may be associated with a venous or arterial thrombotic event: coronary artery disease, coronary vasospasm, myocardial infarction, sudden cardiac death, angina pectoris, unstable angina, cerebrovascular accident, transient ischemic attack, arterial occlusion, deep vein thrombosis, vascular insufficiency, and pulmonary embolus.
The APTC combined endpoint was also reported on.
171 The authors reported that the relative risk of encountering a cardiovascular thrombotic event from taking Vioxx rather than one of the non‑selective NSAIDs used in the analysis was 1.15 (0.63, 2.09). The corresponding figures where the comparator was placebo were 0.94 (0.31, 2.92). The authors concluded:
In summary, an analysis from the rofecoxib osteoarthritis development program found no difference between rofecoxib, comparator nonselective NSAIDs, and placebo in the risks of cardiovascular thrombotic events.
Development of a cardiovascular outcomes study
172 It seems that, during 2001, there was increasing interest within Merck in conducting a large cardiovascular outcomes study with respect to Vioxx. An email of 17 August 2001, tendered by the applicant but not explained by Merck, referred to such a possibility. On 13 September 2001, Dr Scolnick sent an email to some of his colleagues in which he made it clear that MRL would, in the forthcoming period, be able only to carry out studies which were regarded as essential. He left no doubt as to his meaning: “Essential means just that ESSENTIAL. Not preferred, not useful, not helpful; ESSENTIAL.” He proceeded to set out a list of the essential studies, the first item on which read: “For Vioxx: Only the CV outcome study. ONLY ESSENTIAL STUDY!”. There followed some correspondence as to whether it was appropriate to devote limited resources to a single large study at the expense of possibly more useful shorter ones, but Dr Scolnick’s views appeared to attract a measure of support. Under cross‑examination, Dr Reicin was asked about Dr Scolnick’s proposal for a cardiovascular outcomes study. She said that she could not recall him using the word “essential”, but she did recall that he thought that there should be such a study. At the time, there were “multiple conversations with him and all others”.
173 By November 2001, Dr Scolnick’s views on the architecture of a possible cardiovascular outcomes study had advanced somewhat. On 21 November 2001, he sent the following email to a number of his colleagues:
I cannot restrain the activist side of me as I continue to mull the results of the low dose [aspirin] endoscopy study. Looking up Clopidogrel, I think the question we should answer now is NOT whether Vioxx or any Coxib is safe for Cv outcomes. I think the question NOW is what is the best antiplatelet regimen to use with a Coxib. I think that is THE medical question and if we answer it, the problem will dissipate. Thus studies 1. Vioxx low dose [aspirin] and pump inhibitor vs naproxen and pump inhibitor in patients with arthritis and some level of CV risk. 2 Vioxx and clopidogrel vs naproxen and pump inhibitor in same kind of patients. If you think it through, this will answer the questions that are medically needed. Safety committee monitors the studies and stops them if the arms separate statistically with predefined stopping rules. Power for noninferiority based on the incidence and differences seen in our own OA, RA studies.
One of Dr Scolnick’s colleagues replied by asking him whether his email was based on the assumption that that cardioprotective effect of naproxen was medically accepted. I infer that this issue was of concern because Dr Scolnick’s proposal was to include in the study patients with “some level of CV risk”, yet only those in the Vioxx arms would be given low‑dose aspirin. In response to the colleague’s question, Dr Scolnick said (by email): “Our data proves it. We should assume it”.
174 One of the recipients of Dr Scolnick’s original email of 21 November 2001 forwarded it for information to another of her colleagues (with a copy to Dr Reicin) with the comment:
I separately responded that these studies cannot be used as there is no control, and explained that clopidogrel, while it may be an [aspirin] substitute (which naproxen cannot be assumed to be) has not been widely adopted.
Dr Reicin in turn forwarded the email chain to another colleague on 28 November 2001, with the following observation:
[Dr Scolnick] is interested in evaluating whether naproxen is in fact a cardioprotective agent and therefore he is suggesting studies which compare VIOXX + antiplatelet agent versus naproxen without an antiplatelet agent (you assume naproxen is the anti‑platelet agent) in patients who need treatment with aspirin. Since aspirin is the standard of care and does not require steady bid [ie twice daily] dosing to be effective we think it unlikely that physicians would want to take patients off aspirin to put them on naproxen.
175 Senior counsel for the applicant pressed Dr Reicin to concede that this exchange of emails demonstrated that Dr Scolnick wanted to conduct a trial to evaluate whether naproxen was cardioprotective. Dr Reicin did not concede that. In their final submissions, counsel for the applicant maintained that these emails showed that Dr Scolnick was “urging a trial to see whether the naproxen theory was supportable”. I do not so read them. It is true that the respondents did not call Dr Scolnick, but, to the extent that the controversy comes down to an understanding of what he was proposing in his email of 21 November 2001, I think it is tolerably clear that he was concerned to assess the suitability of aspirin and clopidogrel as antiplatelet agents to be used in conjunction with Vioxx. The study design which he had in mind bespeaks (as he said on one of his emails) an assumption that naproxen was cardioprotective.
176 Dr Reicin subsequently worked with her colleagues in MRL to design a study that would test the hypothesis that Vioxx was not prothrombotic and to examine the cardiovascular effects of Vioxx. It seems that there was at least one false start in that endeavour. The applicant tendered a draft protocol titled “A Randomized, Double‑Blind, Parallel, Placebo‑Controlled Trial to Evaluate the Cardiovascular Safety and Efficacy of Rofecoxib on Cardiovascular Events in Patients with Recent Acute Coronary Syndromes” and dated 28 February 2002. The only witness who gave evidence on the subject was Prof Celermajer (one of the cardiologists called by the respondents). He said:
In early 2002, Merck developed a protocol (the VALOR study protocol) which envisaged randomising 10,000 patients to Rofecoxib and 10,000 to receive placebo; all patients would be given low dose aspirin for cardio‑protection as well as a gastric acid inhibitor (Omeprazole) to protect against [gastrointestinal] toxicity. The study population was to comprise patients recently hospitalised with an acute coronary syndrome, with a primary endpoint of CV safety (death from [cardiovascular] or unknown cause, [myocardial infarction], ischaemic stroke or unstable angina requiring hospitalisation).
The VALOR study was eventually not undertaken by Merck, citing three main reasons. Firstly, there was an ethical concern, that the well‑recognised (predominantly renal) effects of Rofecoxib leading to modest elevation of blood pressure and fluid retention might place a post‑acute coronary syndrome (ACS) population at particular risk. Secondly, there were doubts about the generalisability of risk equations from ACS patients to the populations for whom Rofecoxib was principally intended (arthritis patients). Finally, it was thought that the mandated use of aspirin post‑ACS might mask any possible pro‑thrombotic effect of Rofecoxib, should that be present.
Dr Reicin said nothing abut the VALOR study in her evidence. Relevantly, what she said was:
In 2002, I believed that the best way to further test this hypothesis was by designing a study called Protocol 203 for the systematic analysis of cardiovascular safety data pooled from three large scale, long‑term, placebo‑controlled trials of Vioxx 25 mg, that were ongoing or about to get started. There was extensive literature that suggested that COX-2 inhibitors might be able to prevent certain types of cancers or the recurrence of precancerous lesions. Although the three studies comprising Protocol 203 had cancer‑related outcomes as pre‑specified endpoints, Protocol 203 pre‑specified thrombotic cardiovascular safety as its primary hypothesis. MRL designed Protocol 203 in consultation with the FDA, and the protocol was submitted to the FDA on October 10, 2002. After completing a review of the protocol, the FDA responded on December 19, 2002 to questions that Merck had submitted with the protocol on October 10, 2002. [T]he FDA responded that “[a] study with the proposed exposure (size and duration) would provide clinically important safety information about rofecoxib” and that conceptually a meta‑analysis was “reasonable.”
177 On 10 October 2002, MRL forwarded to the FDA a “Request for Special Protocol Assessment”. What was proposed was described as “A Prospective Combined Analysis of Thrombotic Cardiovascular Events in 3 Randomized, Double‑Blind, Placebo‑Controlled Studies of Rofecoxib in Patients at Risk of Developing Recurrent Adenomatous Colon Polyps, Recurrent Colon Cancer, or Prostate Cancer”. Correspondence (and, it seems, a significant teleconference on 4 February 2003) followed between MRL and the FDA. Ultimately, on 25 July 2003 MRL forwarded the details of its new study – known as Protocol 203 – to the FDA. I shall refer to that further below.
The third pooled analysis
178 The third pooled analysis was provided by Merck to the FDA on 22 May 2002. The studies, and the data, included in the analysis were identified as follows:
All Phase IIb through Phase V rofecoxib clinical studies that were at least 4 weeks or more in duration and included a placebo or nonselective NSAID comparator are included in the pooled‑analysis. … Three 6‑week celecoxib studies have been excluded because they did not provide comparisons of rofecoxib to nonselective NSAIDs. Studies are grouped together based on the disease category and these groupings are referred to as study blocks. … The present update includes data from ongoing and new studies up to the data cutoff date of 31‑Jan‑2002. Placebo‑controlled data from the Familial Adenopolyposis (Protocol 129) and Migraine (protocol 125) studies are now available and have been included along with additional data from the ongoing Alzheimer study (protocol 078). The non‑naproxen‑NSAID‑controlled data have been augmented with data from the Arthrotec study (protocol 902). The naproxen‑controlled data have been augmented by data from extensions of the Phase IIb/III rheumatoid arthritis studies (protocols 068, 096, and 097) and from an OA study (Protocol 901). An additional 1043 patients and approximately 2400 patient‑years are included in this updated analysis.
179 The primary endpoint used was, as before, the APTC combined endpoint. However, on this occasion, the analysis used also a secondary endpoint of CVT events. The data were set out in six separate tables, one where each of naproxen, other NSAIDs and placebo was or were the comparator with respect to each of the endpoints.
180 Dealing first with the analyses which used the APTC endpoint, the data showed that the relative risk of taking Vioxx rather than naproxen was 1.63 (1.00, 2.67) for patients in the rheumatoid arthritis studies, 1.55 (0.60, 4.00) for patients in the osteoarthritis studies and 1.61 (1.04, 2.50) for both combined. The APTC data comparing Vioxx with other NSAIDs were derived only from the osteoarthritis studies, and showed an overall relative risk (of taking Vioxx) of 0.87 (0.48, 1.58). The data in the placebo comparison were set out in tabular form, and again I show below only the overall results from each group:
|
Study Block |
Protocol |
Rofecoxib |
Placebo |
Relative Risk (95% CI§) |
|||||
|
N |
Patients With Event/PYR† (Rate‡) |
N |
Patients With Event/PYR† (Rate‡) |
||||||
|
Rofecoxib Versus Placebo |
|||||||||
|
RA |
All |
1622 |
3/338 (0.89) |
989 |
1/201 (0.50) |
1.78 (0.14, 93.68) |
|||
|
OA |
All |
3165 |
12/655 (1.83) |
1215 |
3/232 (1.30) |
1.53 (0.43, 5.44) |
|||
|
Alzheimer |
All |
1453 |
30/1705 (1.76) |
1454 |
41/1934 (2.12) |
0.82 (0.51, 1.32) |
|||
|
Other |
All |
661 |
1/91 (1.10) |
377 |
0/57 (0.00) |
– |
|||
|
Total |
All |
6901 |
46/2788 (1.65) |
4035 |
45/2424 (1.86) |
0.94 (0.62, 1.42) |
|||
|
† Patient-years at risk. ‡ Per 100 PYR § Relative risk of rofecoxib with respect to placebo is calculated from the Cox PH model; if very low total event counts (<11) occur in any block, the ratio of rates with 95% CIs were computed |
|||||||||
181 Turning next to the analyses which used the CVT endpoint, the data showed that the relative risk of taking Vioxx rather than naproxen was 1.85 (1.20, 2.84) for patients in the rheumatoid arthritis studies, 0.90 (0.40, 2.05) for patients in the osteoarthritis studies, and 1.59 (1.09, 2.31) for both combined. A further table broke down the CVT events by sub‑class, but without providing relative risk figures or indications of significance. It was stated that there were, for example, 35 fatal and non‑fatal acute myocardial infarctions in the Vioxx group, within a total of 5943 patient‑years. In the naproxen group, there were 9 such events within 4353 patient‑years. In the case of the “other NSAIDs” CVT comparison, the results were derived only from the osteoarthritis studies, and showed an overall relative risk (of taking Vioxx) of 1.04 (0.65, 1.64). A further table broke down the CVT events by sub‑class, but without providing relative risk figures or indications of significance. It was stated that there were, for example, 14 acute myocardial infarctions in the Vioxx group, within a total of 2687 patient‑years. In the NSAID group, there were 7 such events within 1338 patient‑years.
182 The data in the placebo comparison were set out in tabular form, and again I show only the overall results from each group:
|
Study Block |
Protocol |
Rofecoxib |
Placebo |
Relative Risk (95% CI§) |
|||||
|
N |
Patients With Event/PYR† (Rate‡) |
N |
Patients With Event/PYR† (Rate‡) |
||||||
|
Rofecoxib Versus Placebo |
|||||||||
|
RA |
All |
1622 |
6/337 (1.78) |
989 |
1/201 (0.50) |
3.57 (0.43, 164.3) |
|||
|
OA |
All |
3165 |
20/654 (3.06) |
1215 |
4/232 (1.73) |
1.80 (0.61, 5.30) |
|||
|
Alzheimer |
All |
1453 |
37/1699 (2.18) |
1454 |
48/1925 (2.49) |
0.86 (0.56, 1.33) |
|||
|
Other |
All |
661 |
1/91 (1.10) |
377 |
0/57 (0.00) |
– |
|||
|
Total |
All |
6901 |
64/2782 (2.30) |
4035 |
53/2415 (2.19) |
1.04 (0.72, 1.52) |
|||
|
† Patient-years at risk. ‡ Per 100 PYR § Relative risk of rofecoxib with respect to placebo is calculated from the Cox PH model; if very low total event counts (<11) occur in any block, the ratio of rates with 95% CIs were computed. Includes investigator reported events for studies which were not subject to adjudication (068 and 069-OA). |
|||||||||
A further table broke down the CVT events by sub‑class, but without providing relative risk figures or indications of significance. It was stated that there were, for example, 23 fatal and non‑fatal acute myocardial infarctions in the Vioxx group, within a total of 2782 patient‑years. In the placebo group, there were 18 such events within 2415 patient‑years. The respondents pointed to the circumstance that, in the analysis of 22 May 2002, it was only under the naproxen comparison that there was a statistically significant relative risk of taking Vioxx.
183 In the text of the third pooled analysis, the data were discussed in the following terms:
The earlier pooled‑analysis demonstrated that there was no evidence to suggest that the risk of thrombotic cardiovascular events in patients taking rofecoxib was dissimilar to the risks in patients receiving placebo or the non‑naproxen NSAIDs ibuprofen, diclofenac, and nabumetone. This update reaffirms those previous observations. For both the APTC endpoint and the Thrombotic Cardiovascular Serious Adverse experiences, the risk for rofecoxib relative to placebo and to the non‑naproxen NSAIDs approximated 1 and the 95% CI of the relative risks included 1. It is known that diclofenac, ibuprofen, and nabumetone do not provide sustained and nearly complete (>90%) inhibition of platelet function throughout the dosing interval. Therefore, it is not surprising that there is no meaningful difference between rofecoxib and these non‑naproxen NSAIDs with regard to risk of thrombotic events.
The updated pooled‑analysis included additional naproxen controlled data from both rheumatoid arthritis and osteoarthritis studies. For the APTC combined endpoint, the relative risk for rofecoxib relative to naproxen was elevated in both the rheumatoid arthritis (1.63) and osteoarthritis (1.55) protocols. The overall relative risk was 1.61 (95% CI 1.04, 2.50). These data can be interpreted as representing a 61% greater risk of an APTC event on rofecoxib (95% CI 4, 250%) or a 38% reduction in APTC risk with naproxen compared to rofecoxib (95% CI 4, 60%). Of note, the magnitude of the risk reduction for naproxen relative to rofecoxib is consistent with the overall 25% reduction in the APTC combined endpoint for aspirin compared to placebo in a recent large meta‑analysis (Reference 2). Overall, the data are consistent with the hypothesis that, in these studies, naproxen had a clinically meaningful antiplatelet effect.
….
The results of these updated pooled‑analysis [sic] are consistent with and extend those of previous pooled analyses to now encompass a period of up to 3 years of follow‑up in approximately 30,500 patients representing approximately 18,500 patient-years of experience. The totality of the data in this pooled analysis is most consistent with naproxen having provided a relative cardioprotective benefit in these studies and argue against a prothrombotic effect of rofecoxib.
The Weir article
184 A third article on this general subject was submitted to the American Heart Journal on 13 May 2002, and published as Weir (2003). The authors were Matthew R Weir MD and Drs Sperling, Reicin and Gertz. Their article contained an explanation of the mechanism hypothesized by FitzGerald and a review of the available literature. They stated that the cardiovascular effects of selective COX-2 inhibition were complex, and that understanding of them continued rapidly to evolve. Referring to previous research, they said: “The possibility of a neutral, harmful, or even beneficial effect have all been raised.”
185 The Weir article provided updated data with respect both to Konstam (2001) and to Reicin (2002) article. As to the former, APTC combined endpoint data were presented that were the same as those provided to the FDA on 22 May 2002. The CVT data from the Alzheimer’s studies contained in the Safety Update Report provided to the FDA on 12 July 2001 (see para 166 above) were also set out. As to the Reicin article, the authors added “data from long‑term extensions over 2 years”. That addition altered the NSAID comparator figure referred to in para 171 above to 1.01 (0.59, 1.77). The placebo comparator figure was also altered – to 1.06 (0.34, 3.23) – but, as Prof Woodward pointed out, the data referred to were the same as those relied upon in Reicin (2002). The conclusions expressed in the article included the following:
CV safety information from the rofecoxib program has been carefully reviewed. Rofecoxib has been shown to have little or no effects on platelet aggregation or on platelet‑derived thromboxane synthesis, but it partially reduced systemic prostacyclin synthesis. In the OA phase IIB/III program, similar rates of CV thrombotic events were reported with rofecoxib, placebo, and comparator, nonselective NSAIDs (ibuprofen, diclofenac, and nabumetone). In the VIGOR trial, naproxen (an NSAID with sustained antiplatelet effects throughout its dosing interval) was associated with a significantly lower risk of CV events than rofecoxib. The subsequently conducted pooled analysis from 23 studies representing more than 14,000 patient‑years at risk demonstrated that rofecoxib was not associated with excess CV thrombotic events compared with either placebo or nonnaproxen NSAIDs. Again, naproxen appeared to be the outlier, suggesting a cardioprotective benefit of naproxen.
The fourth pooled analysis
186 The fourth pooled analysis was provided by Merck to the FDA on 22 March 2004. It followed closely the format of the third, and there were new or additional data in relatively minor respects only. The studies, and the data, included in the analysis were identified as follows:
All Phase IIb through Phase V rofecoxib clinical studies that were at least 4 weeks or more in duration and included a placebo or non‑selective NSAID comparator are included in the pooled‑analysis. … Four 6‑week celecoxib studies have been excluded because they did not provide comparisons of rofecoxib to nonselective NSAIDs. Studies in healthy subjects or juveniles were excluded because of the substantially different patient population (e.g., differences in underlying cardiovascular risk factors). Studies are grouped together based on the disease category and these groupings are referred to as study blocks.
…
The present update includes data from ongoing and new studies up to the data cut‑off date of 10‑Jun‑2003. Additional exposure and event data primarily came from the recently completed Alzheimer study (Protocol 078). The ibuprofen and placebo controlled Low Dose Aspirin Study (Protocol 136) has also been available and added to the combined analyses. No meaningful additional data have been available from any other studies; however, due to updates on the database, negligible additions were available on the patient‑years exposure in some studies. An additional 750 patient‑years have been included in this updated analysis. There were 14 new first‑events in the Antiplatelet Trialists’ Collaboration … endpoint and 17 new first events in the Thrombotic Cardiovascular Serious Adverse Experiences … endpoint since the last update of January 2002.
The data were set out in six separate tables, one where each of naproxen, other NSAIDs and placebo was or were the comparator with respect to each of the endpoints. The naproxen data were unchanged from the third analysis. In the case of the “other NSAIDs” APTC comparison, the results were derived only from the osteoarthritis studies, and showed an overall relative risk (of taking Vioxx) of 0.93 (0.51, 1.69). The data in the placebo comparison were set out in tabular form, and may be summarised, by groups, as follows:
|
Study Block |
Study No. |
Rofecoxib |
Placebo |
Relative Risk§ (95% CI) |
||||
|
N |
Cases/PYR† |
Rate‡ (95% CI) |
N |
Cases/PYR† |
Rate‡ (95% CI) |
|||
|
RA |
PN 068 |
332 |
0/49.17 |
0.00 (0.00, 7.50) |
168 |
0/24.46 |
0.00 (0.00, 15.08) |
1.78 (0.14, 93.67) |
|
PN 096 |
459 |
3/97.07 |
3.09 (0.64, 9.03) |
301 |
0/58.02 |
0.00 (0.00, 6.36) |
||
|
PN 097 |
612 |
0/136.55 |
0.00 (0.00, 2.70) |
299 |
0/62.14 |
(0.00 (0.00, 5.94) |
||
|
098+103 |
219 |
0.54.87 |
0.00 (0.00, 6.72) |
221 |
1/56.21 |
1.78 (0.05, 9.91) |
||
|
All RA |
1622 |
3/337.66 |
0.89 (0.18, 2.60) |
989 |
1/200.83 |
0.50 (0.01, 2.77) |
||
|
OA |
PN 029 |
378 |
2/45.64 |
4.38 (0.53, 15.83) |
145 |
0/16.32 |
0.00 (0.00, 22.61) |
2.39 (0.68, 8.41) |
|
PN 033 |
446 |
0/65.77 |
0.00 (0.00, 5.61) |
69 |
1/9.45 |
10.59 (0.27, 58.99) |
||
|
PN 040 |
486 |
1/72.37 |
1.38 (0.03, 7.70) |
74 |
0/10.67 |
0.00 (0.00, 34.57) |
||
|
PN 044 |
381 |
3/154.06 |
1.95 (0.40, 5.69) |
177 |
0/52.01 |
0.00 (0.00, 7.09) |
||
|
PN 045 |
388 |
0/157.20 |
0.00 (0.00, 2.35) |
194 |
2/61.00 |
3.28 (0.40, 11.84) |
||
|
PN 058 |
174 |
1/21.08 |
4.74 (0.12, 26.43) |
52 |
0/6.14 |
0.00 (0.00, 60.07) |
||
|
PN 083 |
98 |
0/20.95 |
0.00 (0.00, 17.61) |
100 |
0/21.16 |
0.00 (0.00, 17.43) |
||
|
PN 085 |
424 |
1/61.31 |
1.63 (0.04, 9.09) |
208 |
0/27.88 |
0.00 (0.00, 13.23) |
||
|
PN 090 |
390 |
4/56.30 |
7.11 (1.94, 18.19) |
196 |
0/26.92 |
0.00 (0.00 13.70) |
||
|
PN 136 |
399 |
1/95.36 |
1.05 (0.03, 5.84) |
816 |
0/200.79 |
0.00 (0.00 1.84) |
||
|
All OA |
3564 |
13/750.05 |
1.73 (0.92, 2.96) |
2031 |
3/432.34 |
0.69 (0.14, 2.03) |
||
|
ALZ |
PN 078 |
723 |
29/1368.76 |
2.12 (1.42, 3.04) |
728 |
29/1562.58 |
1.86 (1.24, 2.67) |
0.99 (0.65, 1.52) |
|
PN 091 |
346 |
4/292.48 |
1.37 (0.37, 3.50) |
346 |
11/367.16 |
3.00 (1.50, 5.36) |
||
|
PN 126 |
380 |
6/186.01 |
3.23 (1.18, 7.02) |
376 |
5/191.56 |
2.61 (0.85, 6.09) |
||
|
All ALZ |
1449 |
39/1847.25 |
2.11 (1.50, 2.89) |
1450 |
45/2121.30 |
2.12 (1.55, 2.84) |
||
|
OTH |
PN 118 |
102 |
0/14.52 |
0.00 (0.00, 25.41) |
58 |
0/8.42 |
0.00 (0.00, 43.83) |
|
|
PN 120 |
252 |
1/27.72 |
3.61 (0.09, 20.10) |
128 |
0/13.77 |
0.00 (0.00, 26.79) |
||
|
PN 121 |
210 |
0/23.35 |
0.00 (0.00, 15.80) |
100 |
0/10.85 |
0.00 (0.00, 34.00) |
||
|
PN 125 |
89 |
0/23.39 |
0.00 (0.00, 15.77) |
83 |
0/21.73 |
0.00 (0.00, 16.98) |
||
|
PN 129 |
8 |
0/3.47 |
0.00 (0.00, 106.43) |
9 |
0/4.38 |
0.00 (0.00, 84.16) |
||
|
All OTH |
661 |
1/92.44 |
1.08 (0.03, 6.03) |
378 |
0/59.15 |
0.00 (0.00, 6.24) |
||
|
All Combined |
7296 |
56/3027.39 |
1.85 (1.40, 2.40) |
4848 |
49/2813.62 |
1.74 (1.29, 2.30) |
1.14 (0.77, 1.68) |
|
|
OA = Osteoarthritis, RA = Rheumatoid Arthritis, ALZ = Alzheimers Studies, and OTH = Other studies. † Patient-years at risk ‡ Per 100 Patient-Years § Relative risk to Placebo using Cox model where the number of cases is at least 11; otherwise, relative risk is a ratio of rates. |
||||||||
187 In the case of the “other NSAIDs” CVT comparison, the results (again from the osteoarthritis studies only) showed an overall relative risk (of taking Vioxx) of 1.09 (0.69, 1.73). A further table broke down the CVT events by sub‑class, but without providing relative risk figures or indications of significance. It was stated that there were, for example, 14 acute myocardial infarctions in the Vioxx group, within a total of 2782 patient‑years. In the NSAID group, there were 7 such events within 1432 patient‑years. A grouped summary of the placebo results is as follows:
|
Study Block |
Study No. |
Rofecoxib |
Placebo |
Relative Risk§ (95% CI) |
||||
|
N |
Cases/PYR† |
Rate‡ (95% CI) |
N |
Cases/PYR† |
Rate‡ (95% CI) |
|||
|
RA |
PN 068 |
332 |
0/49.05 |
4.08 (0.49, 14.73) |
168 |
0/24.46 |
0.00 (0.00, 15.08) |
3.57 (0.43, 164.23) |
|
PN 096 |
459 |
4/97.01 |
4.12 (1.12, 10.56) |
301 |
0/58.02 |
0.00 (0.00, 6.36) |
||
|
PN 097 |
612 |
0/136.55 |
0.00 (0.00, 2.70) |
299 |
0/62.14 |
(0.00 (0.00, 5.94) |
||
|
098+103 |
219 |
0.54.87 |
0.00 (0.00, 6.72) |
221 |
1/56.21 |
1.78 (0.05, 9.91) |
||
|
All RA |
1622 |
6/337.48 |
1.78 (0.65, 3.87) |
989 |
1/200.83 |
0.50 (0.01, 2.77) |
||
|
OA |
PN 029 |
378 |
3/45.60 |
6.58 (1.36, 19.23) |
145 |
0/16.32 |
0.00 (0.00, 22.61) |
2.84 (0.97, 8.38) |
|
PN 033 |
446 |
2/65.69 |
3.04 (0.37, 11.00) |
69 |
1/9.45 |
10.59 (0.27, 58.99) |
||
|
PN 040 |
486 |
1/72.37 |
1.38 (0.03, 7.70) |
74 |
0/10.67 |
0.00 (0.00, 34.57) |
||
|
PN 044 |
381 |
4/154.02 |
2.60 (0.71, 6.65) |
177 |
0/52.01 |
0.00 (0.00, 7.09) |
||
|
PN 045 |
388 |
3/156.97 |
1.91 (0.39, 5.59) |
194 |
3/60.96 |
4.92 (1.01, 14.38) |
||
|
PN 058 |
174 |
1/21.08 |
4.74 (0.12, 26.43) |
52 |
0/6.14 |
0.00 (0.00, 60.07) |
||
|
PN 083 |
98 |
0/20.95 |
0.00 (0.00, 17.61) |
100 |
0/21.16 |
0.00 (0.00, 17.43) |
||
|
PN 085 |
424 |
1/61.31 |
1.63 (0.04, 9.09) |
208 |
0/27.88 |
0.00 (0.00, 13.23) |
||
|
PN 090 |
390 |
5/56.26 |
8.89 (2.89, 20.74) |
196 |
0/26.92 |
0.00 (0.00 13.70) |
||
|
PN 136 |
399 |
1/95.36 |
1.05 (0.03, 5.84) |
816 |
0/200.79 |
0.00 (0.00 1.84) |
||
|
All OA |
3564 |
21/749.62 |
2.80 (1.73, 4.28) |
2031 |
4/432.29 |
0.93 (0.25, 2.37) |
||
|
ALZ |
PN 078 |
723 |
38/1360.87 |
2.79 (1.98, 3.89) |
728 |
36/1550.97 |
2.32 (1.63,3.21) |
1.03 (0.70, 1.53) |
|
PN 091 |
346 |
4/292.48 |
1.37 (0.37, 3.50) |
346 |
12/366.52 |
3.27 (1.69, 5.72) |
||
|
PN 126 |
380 |
6/185.70 |
3.23 (1.19, 7.03) |
376 |
5/191.47 |
2.61 (0.85, 6.09) |
||
|
All ALZ |
1449 |
48/1839.04 |
2.61 (1.92, 3.46) |
1450 |
53/2108.96 |
2.51 (1.88, 3.29) |
||
|
OTH |
PN 118 |
102 |
0/14.52 |
0.00 (0.00, 25.41) |
58 |
0/8.42 |
0.00 (0.00, 43.83) |
|
|
PN 120 |
252 |
1/27.72 |
3.61 (0.09, 20.10) |
128 |
0/13.77 |
0.00 (0.00, 26.79) |
||
|
PN 121 |
210 |
0/23.35 |
0.00 (0.00, 15.80) |
100 |
0/10.85 |
0.00 (0.00, 34.00) |
||
|
PN 125 |
89 |
0/23.39 |
0.00 (0.00, 15.77) |
83 |
0/21.73 |
0.00 (0.00, 16.98) |
||
|
PN 129 |
8 |
0/3.47 |
0.00 (0.00, 106.43) |
9 |
0/4.38 |
0.00 (0.00, 84.16) |
||
|
All |
661 |
1/92.44 |
1.08 (0.03, 6.03) |
378 |
0/59.15 |
0.00 (0.00, 6.24) |
||
|
All Combined |
7296 |
76/3018.58 |
2.52 (1.98, 3.15) |
4848 |
58/2801.23 |
2.07 (1.57, 2.68) |
1.26 (0.89, 1.78) |
|
|
OA = Osteoarthritis, RA = Rheumatoid Arthritis, ALZ = Alzheimers Studies, and OTH = Other studies. † Patient-years at risk ‡ Per 100 Patient-Years § Relative risk to Placebo using Cox model where the number of cases is at least 11; otherwise, relative risk is a ratio of rates. |
||||||||
A further table broke down the CVT events by sub‑class, but without providing relative risk figures or indications of significance. It was stated that there were, for example, 28 fatal and non‑fatal acute myocardial infarctions in the Vioxx group, within a total of 3019 patient‑years. In the placebo group, there were 19 such events within 2801 patient‑years.
188 The report expressed the following conclusions:
1. The risk of sustaining a thrombotic cardiovascular event is similar in patients treated with rofecoxib, placebo, or non‑naproxen non‑selective NSAIDs which lack potent inhibition of platelet function. The data continue to support the lack of a prothrombotic effect with rofecoxib.
2. The risk of sustaining a thrombotic cardiovascular event is reduced in patients treated with naproxen compared to rofecoxib. This reduction in events with naproxen may be due to its ability to maintain near maximal inhibition of platelet function throughout its dosing interval and is consistent with naproxen having provided cardioprotective effects in these studies.
The respondents submitted that, by reference to the data submitted on 22 March 2004, it remained the case that it was only under the naproxen comparison that there was a statistically significant relative risk of taking Vioxx. Dr Reicin interpreted the results of all relevant studies, including VIGOR, down to the fourth pooled analysis as most consistent with the conclusion that Vioxx was cardioneutral, whereas naproxen was cardioprotective.
Other studies referred to by Dr Reicin
189 Dr Reicin said that Merck undertook various other studies to evaluate the FitzGerald hypothesis. One involved an investigation of whether COX-l or COX-2 was responsible for the production of prostacyclin in the rabbit aorta. Dr Reicin said that the study showed that COX-l was responsible for the production of prostacyclin, and that Vioxx, as a COX-2 selective inhibitor, had no effect on the levels of prostacyclin in the aorta. The study was published as Wong (2001). Dr Reicin said that MRL also conducted a similar study in healthy dogs, with similar results.
190 Dr Reicin said that Merck sponsored a study which measured the bleeding time and prostacyclin and thromboxane production at the site of laceration in healthy human volunteers (Tuleja 2003). According to Dr Reicin, the study concluded that dosing with Vioxx did not affect vascular prostacyclin or bleeding time, and that naproxen had an antithrombotic effect, similar to aspirin. The authors concluded that “[i]n healthy men, naproxen exerts an antithrombotic effect as least as potent as aspirin, whereas rofecoxib does not affect hemostatic balance.” The authors did, however, make the following qualification, not referred to by Dr Reicin in her witness statement:
The population in this study was healthy and free of known cardiovascular risk factors. The results, therefore, should not be extrapolated leniently to patients with atherosclerosis, in whom prostanoids are known to play an important role in homeostasis of cardiovascular system.
Protocol 203
191 As I have mentioned above, planning within MRL for an outcomes study designed to assess the cardiovascular risks, if any, associated with the consumption of Vioxx led to the development of a study known as Protocol 203. That was not a trial in its own right, but an analysis of the cardiovascular outcomes from three studies involving patients who had had cancer. The hypotheses to be tested in the study were:
Primary: Rofecoxib 25 mg once daily will be non‑inferior to placebo in the risk of developing a confirmed thrombotic cardiovascular serious adverse experience (ie, the upper limit of the 95% confidence interval … of the hazard ratio will be less than 1.30).
Secondary: Patients treated with rofecoxib 25 mg once daily will have a decreased risk of developing a confirmed thrombotic cardiovascular serious adverse experience relative to placebo (ie, the upper 95% confidence limit on [hazard ratio] < 1.0 and lower limit < 0.77).
The three studies that were to be included in Protocol 203 were identified in the report to the FDA of 25 July 2003 as follows:
- APPROVe (Protocol 122) A Multicenter, Randomized, Parallel‑Group, Placebo‑Controlled, Double‑Blind Study with In‑House Blinding to Determine the Effect of 156 Weeks of treatment with [Vioxx] on the Recurrence of Neoplastic Polyps of the Large Bowel in patients with a History of Colorectal Adenomas. Enrollment is 2612 patients, the duration of active treatment is 3 years.
- VICTOR (Protocol 145) [Vioxx] in Colorectal Cancer Therapy: Definition of Optimal Regime. Anticipated enrollment is approximately 7000 patients. One group of approximately 3500 patients will be treated for 2 years and the other 3500 patients will be treated for 5 years.
- ViP (Protocol 201) A Double‑Blind, Randomized, Placebo‑Controlled, Multicenter Study to Evaluate the Effect of Rofecoxib In Decreasing the Risk of Prostate Cancer. Anticipated enrollment is 15,000 patients, the duration of active treatment is 6 years.
192 The APPROVe study had commenced in February 2000. A total of 2586 patients with a history of colorectal adenomas were randomised to either rofecoxib 25 mg once daily (1287 patients) or placebo (1299 patients). The VICTOR study had commenced in April 2002. There were 1167 patients in the Vioxx arm and 1160 patients in the placebo arm. The ViP study had only just commenced at the time of the report to the FDA of 25 July 2003. The patients in the study were men with a mean age of 63.6 years – 2369 in the Vioxx arm and 2372 in the placebo arm. Each of these studies was ongoing in September 2004 when the events to which I next refer occurred.
The cardiovascular results of APPROVe and the withdrawal of Vioxx
193 On 17 September 2004 the External Safety Monitoring Board for the APPROVe trial met and reviewed data available to 16 August 2004. The Board noted the existence of significant between‑treatment differences in categories which included APTC events and thromboembolic cardiovascular events (as adjudicated in each case). Those data were supported by an analysis of unadjudicated events and by adverse trends in heart failure, pulmonary oedema and cardiac failure composite outcomes. Also, more patients on treatment B were experiencing stage 2 hypertension during follow‑up. The relative risk of a confirmed APTC event for those on treatment B had grown steadily over time – from a point estimate of 1.2 in May 2003 to a point estimate of 2.2 in August 2004. These figures led the Board to recommend that the Executive Committee for the trial be unblinded to the safety data and that the trial itself be discontinued. That recommendation was accepted. After a number of conferences over the next few days involving experts both internal and external to Merck, Dr Peter Kim, the President of MRL, recommended to Mr Raymond Gilmartin, then CEO of Merck, that the best course would be to withdraw Vioxx from the market. That recommendation was also accepted. As a result of these events, the VICTOR and ViP studies were also terminated and Protocol 203 did not run to its intended conclusion.
194 The Clinical Study Report for APPROVe, submitted to the FDA on 6 June 2005, contained as an appendix a Cardiovascular Safety Report dated 15 March 2005. The report set out a table of the confirmed CVT events occurring while patients were on their treatment and up to 14 days after the last dose of study therapy. An edited version of that table is as follows:
Analysis of Confirmed Thrombotic Cardiovascular Events by Class of Terms
(Events on Treatment through 14 Days After the Last Dose of Study Therapy)
|
Event Terms |
Rofecoxib 25 mg (N 1287) |
Placebo (N 1299) |
Comparison |
||||
|
Events Patient-Years† |
PY Rate‡ (95% CI) |
Events Patient-Years† |
PY‑Rate‡ (95% CI) |
Difference (95% CI) |
Relative Risk (95% CI) |
P-Value |
|
|
Total number of patients with events |
46/3059 |
1.50 (1.10, 2.01) |
26/3327 |
0.78 (0.51, 1.15) |
0.72 (0.19, 1.25) |
1.92 (1.19, 3.11) |
0.008 |
|
Cardiac Events |
31/3065 |
1.01 (0.69, 1.44) |
12/3335 |
0.36 (0.19, 0.63) |
0.65 (0.24, 1.06) |
2.80 (1.44, 5.45) |
0.002 |
|
Acute myocardial infarction |
21/3075 |
0.68 (0.42, 1.04) |
9/3338 |
0.27 (0.12, 0.51) |
0.41 (0.07, 0.75) |
2.53 (1.11, 6.28) |
0.024 |
|
Cerebrovascular Events |
15/3080 |
0.49 (0.27, 0.80) |
7/3341 |
0.21 (0.08, 0.43) |
0.28 (-0.01, 0.57) |
2.32 (0.89, 6.74) |
0.091 |
|
Ischemic cerebrovascular stroke |
11/3082 |
0.36 (0.18, 0.64) |
6/3341 |
0.18 (0.07, 0.39) |
0.18 (-0.08, 0.43) |
1.99 (0.67, 6.55) |
0.255 |
|
Peripheral Vascular Events |
3/3087 |
0.10 (0.02, 0.28) |
7/3341 |
0.21 (0.08, 0.43) |
-0.11 (-0.30, 0.08) |
0.46 (0.08, 2.03) |
0.412 |
|
Note: Patients with multiple events may be counted more than once in different terms, but only once in each term. ‡ Events per 100 Patient‑Years |
|||||||
With respect to APTC events, the relative risk of taking Vioxx rather than placebo was 2.06 (1.16, 3.64); p = 0.013. The discussion in the report included the following:
The population enrolled in this study was comprised solely of patients with a previous history of adenomatous polyps. However, with respect to cardiovascular disease they appear to be generally representative of the population at large, including patients with one or more risk factors for CV events and some with a previous history of thrombotic CV events. … Rofecoxib therapy was associated with an increased risk of thrombotic cardiovascular events, in this study. Rofecoxib was associated with a significantly increased risk of sustaining a non‑fatal myocardial infarction, and there was a numeric increase in thrombotic strokes.
195 In his report, the biostatistician called by the respondents, Dr Ronald Marks, referred to an extended analysis by Merck, dated 26 May 2006, of the data from the APPROVe trial. It looked at the data in various ways: (1) by including confirmed CVT and APTC events and all‑cause deaths, occurring in the period up to the 210th week after the patient concerned commenced study therapy (ie the 3‑year treatment period plus a further year); (2) by including all such events, and deaths, occurring down to 31 October 2005; (3) by including results during the follow‑up for patients who were on treatment for at least 150 weeks; (4) by including results during the follow‑up for various (specified) periods of time; and (5) by including confirmed CVT and APTC events, and all‑cause deaths, but excluding patients who had had a CVT or an APTC event, respectively (or, in the case of deaths, either of such events) within the base study period (ie up to the 14th day after discontinuation of treatment). In his report, Dr Marks drew upon data from tables in this analysis which recorded events which had occurred after the base study period and up to the 210th week after the commencement of study therapy by the patient concerned (ie a subset of category (1)). He showed only the rates (per 100 patient years) of events in each of the arms of the study; he did not show relative risk.
196 In the Merck analysis of 26 May 2006, the table of CVT events corresponding to that which I have set out in para 194 above and which dealt with what I have called category (1) events was the following (similarly edited):
Analysis of Confirmed TCVSAE Endpoint by Class of Terms
All Patients (ITT Population) and censored at Week 210
|
Event Terms |
Rofecoxib 25 mg (N=1287) |
Placebo (N=1299) |
Comparison |
|||
|
Events Patient-Years† |
PY Rate‡ (95% CI) |
Events Patient-Years† |
PY‑Rate‡ (95% CI) |
Relative Risk§ (95% CI) |
p-Value |
|
|
Total number of patients with events |
71/4557 |
1.56 (1.22, 1.97) |
42/4697 |
0.89 (0.64, 1.21) |
1.74 (1.19, 2.55) |
0.0043 |
|
Cardiac Events |
45/4590 |
0.98 (0.72, 1.31) |
28/4725 |
0.59 (0.39, 0.86) |
1.65 (1.03, 2.65) |
0.0367 |
|
Fatal/Non-Fatal Acute myocardial infarction |
31/4601 |
0.67 (0.46, 0.96) |
15/4726 |
0.32 (0.18, 0.52) |
2.12 (1.15, 3.93) |
0.0168 |
|
Peripheral Vascular Events |
6/4627 |
0.13 (0.05, 0.28) |
8/4731 |
0.17 (0.07, 0.33) |
0.77 (0.27, 2.21) |
0.6228 |
|
Fatal/Non-Fatal Pulmonary embolism |
3/4631 |
0.06 (0.01, 0.19) |
2/4740 |
0.04 (0.01, 0.15) |
1.54 (0.18, 18.38) |
0.9782 |
|
Cerebrovascular Events |
23/4606 |
0.50 (0.32, 0.75) |
7/4733 |
0.15 (0.06, 0.30) |
3.38 (1.45, 7.88) |
0.0048 |
|
Fatal/Non-Fatal Ischemic cerebrovascular stroke |
17/4614 |
0.37 (0.21, 0,59) |
6/4736 |
0.13 (0.05, 0.28) |
2.91 (1.15, 7.39) |
0.0244 |
|
† Patient-years at risk ‡ Patients with events per 100 Patient-Years § Relative risk to Placebo using Cox model where the number of cases is at least 11; otherwise, relative risk is a ratio of rates. Note: Patients with multiple events may be counted more than once in different terms, but only once in each item. |
||||||
The corresponding table for category (2) events was the following (also similarly edited):
Analysis of Confirmed TCVSAE Endpoint by Class of Terms
All Patients (ITT Population) and censored at 31‑Oct‑2005
|
Event Terms |
Rofecoxib 25 mg (N=1287) |
Placebo (N=1299) |
Comparison |
|||
|
Events Patient-Years† |
PY Rate‡ (95% CI) |
Events Patient-Years† |
PY‑Rate‡ (95% CI) |
Relative Risk§ (95% CI) |
p-Value |
|
|
Total number of patients with events |
76/5081 |
1.50 (1.18, 1.87) |
46/5236 |
0.88 (0.64, 1.17) |
1.70 (1.18, 2.46) |
0.0044 |
|
Cardiac Events |
49/5125 |
0.96 (0.71, 1.26) |
32/5270 |
0.61 (0.42, 0.86) |
1.57 (1.01, 2.46) |
0.0461 |
|
Fatal/Non-Fatal Acute myocardial infarction |
34/5138 |
0.66 (0.46, 0.92) |
18/5274 |
0.34 (0.20, 0.54) |
1.94 (1.09, 3.43) |
0.0232 |
|
Peripheral Vascular Events |
6/5172 |
0.12 (0.04, 0.25) |
8/5283 |
0.15 (0.07, 0.30) |
0.77 (0.27, 2.21) |
0.6228 |
|
Fatal/Non-Fatal Pulmonary embolism |
3/5178 |
0.06 (0.01, 0.17) |
2/5293 |
0.04 (0.00, 0.14) |
1.53 (0.18, 18.36) |
0.9795 |
|
Cerebrovascular Events |
24/5145 |
0.47 (0.30, 0.69) |
7/5284 |
0.13 (0.05, 0.27) |
3.52 (1.52, 8.18) |
0.0034 |
|
Fatal/Non-Fatal Ischemic cerebrovascular stroke |
18/5156 |
0.35 (0.21, 0.55) |
6/5288 |
0.11 (0.04, 0.25) |
3.08 (1.22, 7.76) |
0.0170 |
|
† Patient-years at risk ‡ Patients with events per 100 Patient-Years § Relative risk to Placebo using Cox model where the number of cases is at least 11; otherwise, relative risk is a ratio of rates. Note: Patients with multiple events may be counted more than once in different terms, but only once in each item. |
||||||
The table in the Merck analysis from which Dr Marks’ figures were drawn was the following (also similarly edited; I have emphasised the figures actually cited by Dr Marks in his report):
Analysis of Confirmed TCVSAE Endpoint by Class of Terms
During Follow-up (> 14 days from last does of base study therapy) and Censored at Week 210
|
Event Terms |
Rofecoxib 25 mg (N=1081) |
Placebo (N=1097) |
Comparison |
|||
|
Events Patient-Years† |
PY Rate‡ (95% CI) |
Events Patient-Years† |
PY‑Rate‡ (95% CI) |
Relative Risk§ (95% CI) |
p-Value |
|
|
Total number of patients with events |
28/1535 |
1.82(1.21, 2.64) |
16/1402 |
1.14 (0.65, 1.85) |
1.64 (0.89, 3.04) |
0.1145 |
|
Cardiac Events |
16/1543 |
1.04 (0.59, 1.68) |
16/1402 |
1.14 (0.65, 1.85) |
0.95 (0.47, 1.90) |
0.8850 |
|
Fatal/Non-Fatal Acute myocardial infarction |
10/1540 |
0.65 (0.31, 1.19) |
6/1399 |
0.43 (0.16, 0.93) |
1.67 (0.61, 4.59) |
0.3232 |
|
Peripheral Vascular Events |
3/1544 |
0.19 (0.04, 0.57) |
0/1401 |
0.00 (0.00, 0.26) |
– (0.37, ∞) |
0.2883 |
|
Fatal/Non-Fatal Pulmonary embolism |
3/1544 |
0.19 (0.04, 0.57) |
0/1401 |
0.00 (0.00, 0.26) |
– (0.37, ∞) |
0.2883 |
|
Cerebrovascular Events |
9/1542 |
0.58 (0.27, 1.11) |
0/1401 |
0.00 (0.00, 0.26) |
– (1.79, ∞) |
0.0060 |
|
Fatal/Non-Fatal Ischemic cerebrovascular stroke |
7/1542 |
0.45 (0.18, 0.94) |
0/1401 |
0.00 (0.00, 0.26) |
– (1.31, ∞) |
0.0217 |
|
† Patient-years at risk ‡ Patients with events per 100 Patient-Years § Relative risk to Placebo using Cox model where the number of cases is at least 11; otherwise, relative risk is a ratio of rates. Note: Patients with multiple events may be counted more than once in different terms, but only once in each item. |
||||||
197 As will be observed, the events in these tables do not precisely line up with those in the table in para 194. However, a comparison of the relative risk figures only for the more important events may be made as follows (in which I have included the APTC events as reported):
|
Event terms |
Base Study Period |
To 31 Oct 05 |
To 210th week |
Follow-up only |
|
CVT events |
1.92 (1.19, 3.11) |
1.70 (1.18, 2.46) |
1.74 (1.19, 2.55) |
1.64 (0.89,3.04) |
|
Cardiac events |
2.80 (1.44, 5.45) |
1.57 (1.01, 2.46) |
1.65 (1.03, 2.65) |
0.95 (0.47, 1.90) |
|
Acute myocardial infarction |
2.53 (1.11, 6.28) |
1.94 (1.09, 3.43) |
2.12 (1.15, 3.93) |
1.67 (0.61, 4.59) |
|
Cerebrovascular events |
2.32 (0.89, 6.74) |
3.52 (1.52, 8.18) |
3.38 (1.45, 7.88) |
- |
|
Ischemic Cerebrovascular stroke |
1.99 (0.67, 6.55) |
3.08 (1.22, 7.76) |
2.91 (1.15, 7.39) |
- |
|
Peripheral vascular events |
0.46 (0.08, 2.03) |
0.77 (0.27, 2.21) |
0.77 (0.27, 2.21) |
- |
|
APTC events |
2.06 (1.16, 3.64) |
1.79 (1.17, 2.73) |
1.86 (1.19, 2.90) |
1.64 (0.81, 3.35) |
198 The cardiovascular results of the APPROVe trial were published by a group of authors, including all the members of the Steering Committee, as Bresalier (2005). Their conclusion was: “Among patients with a history of colorectal adenomas, the use of rofecoxib was associated with an increased cardiovascular risk.”
199 About three years later, substantially the same group of authors published the extended results of the trial in Baron (2008) (Dr Baron himself having been the chair of the Steering Committee). They used the data down to 31 October 2005 (ie what I have described in para 195 above as category (2)). They expressed the following conclusion:
Despite limitations of the available data, we have shown that rofecoxib increases the risk of cardiovascular events such as myocardial infarction and stroke. … The cardiovascular toxicity seems to be a class effect; indeed, studies of other selective COX-2 inhibitors have reported similar findings. Conventional non‑aspirin NSAIDs may share the same toxicity to the extent that they are COX-2 selective. All these drugs are effective analgesic and anti‑inflammatory agents, and seem to reduce risks of colorectal neoplasia. However, these benefits will have to be weighed against their proven or possible cardiovascular risks in assessing their suitability in various clinical settings.
As to the reasons why this risk might arise, the authors said:
The mechanisms underlying the cardiovascular effects of rofecoxib and other COX-2 inhibitors are not clear, but several possibilities have been proposed. [Fn: Grosser T, Fries S, FitzGerald GA. “Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities.” Clin Invest 2006; 116: 4‑15.] Side effects of rofecoxib include fluid retention and increased blood pressure, both of which may trigger cardiovascular events. [Fn: Zhang J, Ding EL, Song Y. “Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta‑analysis of randomized trials.” AMA 2006; 296: 1619‑32; and Aw TJ, Haas SJ, Liew D, Krum H. “Meta‑analysis of cyclooxygenase‑2 inhibitors and their effects on blood pressure.” Arch Intern Med 2005; 165: 490‑96.] Also, COX-2 inhibition reduces production of PGI2 by the vascular endothelium, which may lead to increased cardiovascular risk. [Fn: Grosser et al] Finally, COX-2 inhibition may promote atherogenesis or destabilise atherosclerotic plaques. [Fn: Grosser et al]
part iii − vioxx in australia
The statutory and regulatory framework
200 Vioxx tablets, and rofecoxib (as an ingredient used in the manufacture of Vioxx tablets), were “therapeutic goods” within the meaning of the Therapeutic Goods Act 1989 (Cth) (“the TG Act”). Under s 20 of the TG Act (as it stood on the introduction of Vioxx in 1999), it was an offence intentionally to import, to manufacture or to supply therapeutic goods for use in humans unless (relevantly to the present case) such goods were included in the ARTG maintained pursuant to s 17. By s 23(2) of the TG Act, an application for registration was not “effective” unless the prescribed fee had been paid, the applicant had delivered such reasonable number of samples as the Secretary required, and –
… the applicant has delivered to the office to which the application was made such information, in a form approved, in writing, by the Secretary, as will allow the determination of the application ….
A regular application for registration having been made, the goods were to be “evaluated for registration” having regard to circumstances which included the following, listed in s 25(1) of the TG Act:
(d) whether the quality, safety and efficacy of the goods for the purposes for which they are to be used have been satisfactorily established; and
(e) whether the presentation of the goods is acceptable; and
(f) whether the goods conform to any standard applicable to the goods, or any requirements relating to advertising applicable under the regulations; and
(g) if a step in the manufacture of the goods has been carried out outside Australia—whether the manufacturing and quality control procedures used in the manufacture of the goods are acceptable; and
(h) if the goods have been manufactured in Australia—whether the goods have been manufactured in accordance with Part 4; and
(i) whether the goods contain substances that are prohibited imports for the purposes of the Customs Act 1901; and
(j) such other matters (if any) as the Secretary considers relevant.
By s 25(4), once the evaluation of the goods had been completed, the Secretary was required to notify the applicant of his or her decision and, relevantly to the present case, to notify the applicant that the goods would be included in the ARTG, include the goods in the ARTG and give the applicant a certificate of registration.
201 The respondents conducted their case by reference to a silent assumption that s 25 required the Secretary to be satisfied that the quality, safety and efficacy of the goods, the subject of an application for registration, for the purposes for which they were to be used had been satisfactorily established. However, s 25 did not so provide. It provided for the goods to be evaluated having regard to identified circumstances, but it did not make the Secretary’s satisfaction as to the existence of each of those circumstances a condition of the exercise of power to register. Notwithstanding that qualification, I accept that the scheme of the TG Act was that the Secretary would generally be satisfied of the existence of those circumstances – including that relating to the quality, safety and efficacy of the goods – before he or she decided to register.
202 By s 28 of the TG Act, where the Secretary included goods in the ARTG, he or she might impose conditions on the registration. By subs (2), those conditions might relate to:
(a) the manufacture of the goods; or
(b) the custody, use, supply, disposal or destruction of the goods; or
(c) the keeping of records relating to the goods; or
(d) matters dealt with in, or matters additional to matters dealt with in, standards applicable to the goods; or
(e) such other matters relating to the goods as the Secretary thinks appropriate.
203 By s 29A of the TG Act, a person in relation to whom therapeutic goods were registered was required to give to the Secretary information of which he or she became aware and which was of the following kinds:
(a) information that contradicts information already furnished by the person under this Act;
(b) information that indicates that the use of the goods in accordance with the recommendations for their use may have an unintended harmful effect;
(c) information that indicates that the goods, when used in accordance with the recommendations for their use, may not be as effective as the application for registration of the goods or information already furnished by the person under this Act suggests.
204 Section 31 of the TG Act gave the Secretary power to require an applicant for the registration of therapeutic goods, or a person in relation to whom therapeutic goods were registered, to provide such information or documents as the Secretary specified. The power could be exercised in relation to the following categories of information:
(a) the formulation of the goods;
(b) the composition of the goods;
(c) the design specifications of the goods;
(d) the quality of the goods;
(e) the method and place of manufacture or preparation of the goods and the procedures employed to ensure that proper standards are maintained in the manufacture and handling of the goods;
(f) the presentation of the goods;
(g) the safety and efficacy of the goods for the purposes for which they are to be used;
(h) the conformity of the goods to a requirement relating to advertising applicable under the regulations;
(j) the regulatory history of the goods in another country;
(k) any other matter prescribed by the regulations for the purposes of this paragraph in relation to goods of that kind.
205 Section 32 of the TG Act dealt with the subject of “Inspection and Variation of Entries in Register”. It was provided that the register was not open for public inspection, but that “a person in relation to whom the therapeutic goods are registered” might make a written request to the Secretary for a copy of the entry in the register in relation to the goods. The next three subsections dealt with the disposition by the Secretary of such a request. Subsection (4) provided as follows:
Where:
(a) the person in relation to whom therapeutic goods are registered or listed has asked the Secretary to vary product information included in the entry in the Register that relates to the goods; and
(b) the only effect of the variation would be to reduce the class of persons for whom the goods are suitable or to add a warning or precaution, being a warning or precaution that does not include any comparison of the goods with any other therapeutic goods by reference to quality, safety or efficacy;
the Secretary must vary the entry in accordance with the request.
By subs (6), “product information” was defined as “information relating to the safe and effective use of the goods, including information regarding the usefulness and limitations of the goods”. The TG Act did not otherwise use, or refer to, the concept of “product information”. The Therapeutic Goods Regulations 1990 assumed that there would be something called “product information”, but did not deal with the subject as such.
206 In July 1994, the TGA published the second edition of the “Australian Guidelines for the Registration of Drugs”, volume one of which dealt with the subject “Prescription and Other Specified Drug Products”. The introductory memorandum to the guidelines stated that they described “the information to be supplied with an application for registration of therapeutic goods”, and that the guidelines were “approved for the purposes of subs 23(2)” of the TG Act. A section of the guidelines dealt with the subject “Specific Australian Administrative Requirements”. One of the items in that section was headed “Product Information (PI)”. The term “product information” was defined simply as follows: “Section 32. Also referred to as Product Information”. As is apparent from what I have set out above, s 32 defined “product information” as information of a certain kind. The TG Act did not, however, directly require that information to be provided, much less in a particular form. The guidelines, however, took those next steps. They gave the term initial capitals and treated it as a reference to a document, of which, for example, there could be a “copy”. They required a copy of the draft Product Information to be submitted by the applicant for registration. It was then said:
The PI is to be regarded as a document which contains information sufficient to ensure safe and effective use of the drug under nearly all circumstances. It is to present a scientific, objective account of the drug’s usefulness and limitations as shown by the data supporting the application. It is to be devoid of promotional material.
207 The guidelines stated that, after registration, the Product Information must not be changed without TGA approval, save in the case of “safety‑related changes” which might be made, and notified to the TGA, at any time, subject to the following conditions:
– the change must only reduce the patient population or add a warning or precaution (this is interpreted to include adding further restrictions to the use of the drug such as deleting an indication, or adding adverse reactions or contraindications); and
– the TGA must be notified of such changes within 5 working days of implementation; and
– the notification must take the form of submitting a copy of the approved product information on which the changes have been made and clearly identified; and
– an accompanying letter should state that no other changes have been made and that the changes are supported by data in the sponsor’s possession; and
– the subsequently printed version of the Product Information must show the date of the lest [sic] TGA approval, if applicable, and the date of the most recent notification …
208 The guidelines identified the information that should be contained in the Product Information. Although lengthy, because of its importance in the present proceeding, I set out that identification as follows:
The PI should contain information under the following headings:
(i) Name of the drug
– the non‑proprietary name of the substance, or in the case of a mixture of substances, of each therapeutically active ingredient;
– chemical structure should generally be included.
(ii) Description
– a description of relevant physical and chemical characteristics of the drug and its formulations. Australian Approved Names should be used;
– excipients should be listed for new product registrations (but may be included under (x) Presentation). Inclusion of excipients is recommended for PI updates.
(iii) Pharmacology
– the pharmacology, including pharmaco‑kinetics and pharmacological actions especially in humans.
(iv) Indications
– the therapeutic applications of the drug, clearly stating whether the treatment is curative, palliative, adjunctive, including any conditions imposed by ADEC or TGA.
(v) Contraindications
– situations where patients should never or generally not be treated with this drug. In rare cases where the drug should never be given, this must be specifically outlined.
(vi) Precautions
Include information under the following subheadings:
– Use with caution in the following circumstances (e.g. describe particular populations or clinical situations where dosage reduction is required)
– Check the following before use (e.g. specify particular investigations that may need to be carried out)
– Carcinogenicity/mutagenicity
– Use in pregnancy
Include a proposed or approved Australian Pregnancy Categorization, any relevant standard text and other information consistent with this categorization.
– Use in lactation
– Interactions with other drugs
Include known clinically relevant interactions and other potentially serious interactions based on the pharmacology of the drug. It is useful to group interactions according to outcome (e.g. potentiation or reduction of effect) and to explain the mechanism of action where this is known.
– Effects on laboratory tests
(vii) Adverse Reactions
– an indication of severity, clinical importance and frequency should be given.
(viii) Dosage and Administration
Where relevant, include information on:
– dosage (dose and interval);
– dosage adjustment in renal or liver insufficiency, dialysis, concomitant disease;
– maximum tolerated daily dose and the maximum dose for an entire course of therapy;
– monitoring advice; and
– other pertinent information such as relationship to meals and compatibility with other drugs and fluids.
(ix) Overdosage
– the symptoms, signs and recommended treatment of overdosage or accidental poisoning.
(x) Presentation
– the presentation, including dosage form and quantity, proportion or strength of each therapeutically active ingredient.
(xi) Name and address of the sponsor
209 Thus the Product Information was given a formal documentary status by the 1994 guidelines. It is important to note, however, that this arose under the Secretary’s authority to approve the form in which information should be provided under s 23 of the TG Act, rather than by force of any provision of that Act itself.
210 A series of amendments was made to the TG Act by the Therapeutic Goods Amendment (Medical Devices) Act 2002 (Cth), taking effect on 4 October 2002. At least to the extent relevant for present purposes, there was no amendment of any substance. However, I note that ss 17 and 32 were repealed, and a new Ch 2 headed “Australian Register of Therapeutic Goods” was inserted after s 9. What had been s 17 became (with substantial amendments not presently relevant) s 9A, and what had been s 32 was (also substantially amended) divided into the new ss 9C and 9D. The previous subss (4) and (6) of s 32 were now subss (2) and (5) of s 9D.
Registration of Vioxx on the ARTG
211 Evidence as to the registration of Vioxx in Australia was given by Mr Warren Back, who, from October 1997 to September 1999 was employed by MSDA as a Regulatory Affairs Associate in the Human Health Division, reporting to the then Manager of Regulatory Affairs, Judith Griffin. At that time, his primary role involved the preparation and filing of regulatory submissions to the TGA for new products, including Vioxx. In September 1999, he was appointed as Senior Regulatory Affairs Associate at MSDA, where the responsibilities for his product portfolio included directly managing Australian regulatory submissions to the TGA and regulatory compliance, and acting as MSDA’s point of contact with the TGA. He continued to be responsible for Vioxx from a regulatory perspective, along with Ms Griffin. From January 2003 to December 2004, Mr Back was employed as Regulatory Affairs Manager at MSDA, where he was responsible for overall management of the Regulatory Affairs team and overseeing regulatory submissions and regulatory compliance.
212 MSDA submitted its application for the registration of Vioxx in the TGA on 22 December 1998. The TGA acknowledged receipt on 8 January 1999 and, after making, and receiving a response to, one s 31 request, formally accepted the application for evaluation on 8 February 1999. The indications for which MSDA sought registration of Vioxx were the acute and chronic treatment of the signs and symptoms of osteoarthritis, the relief of pain and the treatment of dysmenorrhoea. The supporting material, of which there were 129 volumes, included:
· in Part I, a draft of each of the proposed Product Information, the proposed Consumer Medicine Information, the proposed text to be included in the labelling for the product and details of the overseas regulatory status of the product;
· in Part II, chemical and pharmaceutical documentation relating to the synthetic process for rofecoxib, the formulation development of Vioxx, the manufacturing process of Vioxx, finished product release testing of Vioxx, stability testing of Vioxx and bioavailability and bioequivalence studies conducted in connection with Vioxx;
· in Part III, pharmacotoxicological (pre-clinical) documentation, including both in vitro test data and data in animal species; and
· in Part IV, clinical documentation, which included data from pharmacokinetic studies, bioavailability and bioequivalence studies, and safety and efficacy studies in humans.
213 MSDA submitted expert reports as follows:
· on the chemical and pharmaceutical documentation, signed by Dr Meredith Cotton, the Director of Technical Affairs, Pharmaceutical Research & Development, Merck Frosst Centre for Therapeutic Research, Quebec, Canada;
· on the pharmacotoxicological documentation, signed jointly by Dr Chi‑Chung Chan, Senior Investigator, Pharmacology, Merck Frosst Centre, and Dr Kamlesh Vyas, Director, Drug Metabolism Administration, Merck Research Laboratories, West Point, PA, USA;
· on the clinical documentation, signed by A/Prof Désirée van der Heijde, Associate Professor in Rheumatology, University Hospital Maastricht, the Netherlands;
· (included in A/Prof van der Heijde’s report) an assessment of the gastrointestinal safety program signed by Prof Christopher Hawkey, Professor of Gastroenterology, University Hospital Nottingham UK, and an assessment of the renal effects of rofecoxib signed by Prof D Craig Barter, Professor and Chairman, Department of Medicine, Indiana University School of Medicine, Indianapolis, Indiana, USA.
The application for registration included data from clinical trials involving patients who were exposed to Vioxx or comparators for periods of up to 86 weeks. According to Mr Back, the clinical trial data met the European Guidelines entitled “Points to Consider on the Investigation of Medicinal Products used in the Treatment of Osteoarthritis”, adopted by the TGA in July 1998.
214 The TGA made three s 31 requests in February and March 1999 in connection with the Vioxx application. MSDA responded to each of these. The TGA issued its first evaluation report on 12 April 1999, which related to matters of chemistry, manufacturing and quality control. In connection with that, the TGA made two further s 31 requests, on 13 April and 19 May 1999. After they were responded to, a further s 31 request was made on 6 August 1999 and responded to on 16 August. The TGA cleared the chemistry and quality control aspects of the application on 19 August 1999.
215 The clinical evaluation report of the TGA was received by MSDA on 16 June 1999. The purpose of such a report was to address whether the clinical data supported the therapeutic indications requested by the sponsor. Here the clinical evaluator recommended that Vioxx at the proposed doses be approved for the treatment of osteoarthritis, but not be approved for the relief of pain, or for the treatment of primary dysmenorrhoea. On 23 July 1999, MSDA submitted a response to the clinical evaluator’s recommendations, stating that it believed that the evaluator had not understood the clinical significance of the data submitted in the application for the acute pain and dysmenorrhoea indications, and had confused the statistical power for the study protocol for the statistical significance of the results.
216 On 12 August 1999, the TGA made a s 31 request in relation to its evaluation of the Vioxx pre-clinical data (safety studies involving animals conducted prior to the commencement of studies in humans). MSDA responded on 19 August 1999. The TGA issued a pre-clinical evaluation report on 20 August 1999, which contained certain recommendations as to bone marrow toxicity, as to further studies of embryofetal development in rabbits and some other animal studies, and with respect to a particular modification to the Product Information. None of these was considered to stand in the way of registration. That was the final evaluation report by the TGA to be provided to MSDA.
217 Dr Philip Chipman was the delegate of the Secretary of the Department of Health and Family Services in matters relevant to the Vioxx registration application. He issued a “delegate’s overview” on 26 August 1999, which summarised each of the evaluation reports theretofore issued, and set out his proposed action with respect to the registration of Vioxx. Dr Chipman proposed to approve Vioxx for the symptomatic treatment of osteoarthritis, but, in the absence of evidence supporting 24‑hour pain relief, to reject the acute pain and dysmenorrhoea indications. However, he also requested advice from the Australian Drug Evaluation Committee (“ADEC”), a specialist committee to advise the TGA as to the approval of prescription medicines for inclusion in the ARTG.
218 MSDA had, and took, the opportunity to respond to the delegate’s overview by submitting a “pre‑ADEC response”. Mr Back prepared that response, which was submitted on 10 September 1999. Following the required format, it included the response as such (which restated MSDA’s position as to the suitability of Vioxx for acute pain and dysmenorrhoea), line listings of serious unexpected adverse drug reactions not mentioned in the proposed Product Information that had not been previously submitted to the TGA, the proposed Product Information and Consumer Medicine Information, a statement of the international regulatory status of Vioxx at the time and the then approved equivalent Product Information for the USA and the UK.
219 There was a pharmaceutical sub‑committee, the role of which was to make recommendations to ADEC on the assessment of the chemistry and manufacturing details of applications. The sub‑committee considered the Vioxx application at its meeting on 27 September 1999 and, on 12 October 1999, recommended approval of Vioxx at dosages of 12.5 mg and 25 mg, while also recommending a number of revisions to the pharmacokinetics section of the Product Information. It had no objection to the approval of Vioxx. The remaining sections of the application would be addressed by ADEC itself.
220 On 7 and 8 October 1999, ADEC considered the Vioxx application, and resolved as follows (as advised to MSDA in correspondence dated 15 October 1999):
1. THERE SHOULD BE NO OBJECTION TO APPROVAL OF THE APPLICATION BY MERCK SHARP & DOHME (AUSTRALIA) PTY LIMITED TO REGISTER VIOXX TABLETS AND ORAL SUSPENSION, CONTAINING ROFECOXIB 12.5 MG AND 25 MG AND 12.5 MG/5 ML AND 25 MG/5 ML, RESPECTIVELY, FOR THE SYMPTOMATIC TREATMENT OF OSTEOARTHRITIS.
THE APPLICATION TO REGISTER VIOXX TABLETS AND ORAL SUSPENSION FOR THE RELIEF OF ACUTE PAIN AND TREATMENT OF PRIMARY DYSMENORRHOEA SHOULD BE REJECTED ON THE GROUNDS OF ABSENCE OF EVIDENCE THAT ANALGESIA CAN BE MAINTAINED FOR 24 HOURS (THE PROPOSED DOSING INTERVAL).
2. APPROVAL SHOULD BE SUBJECT TO FINALISATION OF THE PRODUCT INFORMATION TO THE SATISFACTION OF THE TGA.
Following the ADEC meeting, the TGA required MSDA to take a number of steps with a view to finalising the Vioxx Product Information and Consumer Medicine Information before it was prepared to grant approval.
221 On 20 October 1999, MSDA submitted to the TGA the amendments to the Product Information that had been requested by ADEC and its sub‑committee. This was the subject of a s 31 request made by the TGA on 8 November 1999 which related to several sections in the “Clinical Pharmacology”, “Contraindications”, “Precautions” and “Dosage and Administration” sections of the Product Information. MSDA responded on 11 November 1999, submitting a further revised Product Information for approval. It incorporated the majority of the amendments requested by the TGA, and suggested revised wording in some cases.
222 On 15 December 1999, the TGA made another s 31 request which sought revisions to the section of the Product Information which described the impairment of fertility and pup growth rate seen in the rat studies. On 16 December 1999 there was a telephone conference which involved Dr Chipman, another representative of the TGA, a representative of Merck’s toxicology group, Ms Sandy Simpson (then Associate Director of Regulatory Affairs at Merck and part of the Merck Regulatory Affairs International Group) and Mr Back. They discussed, and agreed on, appropriate wording for the Product Information sections describing the rat studies. MSDA responded to the s 31 request on 17 December 1999, submitting a further revised Product Information. On the same day Mr Back sent a facsimile to the TGA, setting out a revised version of the Consumer Medicine Information in line with revisions to the Product Information.
223 Dr Chipman subsequently required one further terminological amendment to the Product Information, to which Mr Back attended on 21 December 1999. The TGA’s final s 31 request was made on 22 December 1999, and recommended changes to the Consumer Medicine Information that had been submitted earlier that month. On the same day, MSDA submitted a revised Consumer Medicine Information by which it adopted all of the TGA’s requested changes.
224 By letter dated 23 December 1999, Dr Chipman, as delegate of the Secretary, informed MSDA that approval had been granted for the registration of Vioxx tablets (12.5 mg and 25 mg) and oral suspension (12.5 mg/5 ml and 25 mg/5 ml) pursuant to s 28 of the TG Act, but that a number of conditions had been imposed. Of those that presently need to be mentioned, one was that Vioxx was indicated only for the symptomatic treatment of osteoarthritis. Another, under the heading “Product Information”, stated that the text of the Product Information submitted by MSDA (most recently on 17 and 21 December 1999) was “considered satisfactory”. Another was as follows:
The Product Information (reference 4.20 AGRD1, 2nd edition) must meet with the TGA’s approval at all times. With the exception of safety related changes that further restrict the use of the product, which must be notified to the TGA within five working days, any proposed changes to the approved text must be submitted and be approved by the Administration prior to distribution.
The safety related changes referred to above are defined as those that delete an indication or reduce the patient population, or add a warning, precaution, adverse reaction or contraindication.
The reference to para 4.20 of “AGRD1” was a reference to the 1994 guidelines to which I have referred at para 206 above. Another condition was that promotional material, other than the Product Information, had to comply with the requirements of the Code of Conduct of the Australian Pharmaceutical Manufacturers Association. It was also required that MSDA submit post-marketing reports, meeting the requirements for “periodic safety update reports”, not less frequently than annually for the succeeding three years, the first being submitted no later than 15 months after the date of the approval letter. Registration certificates issued on 17 January 2000. The registration of Vioxx was effective from this date.
225 The original Product Information (approved on 23 December 1999) contained nothing to suggest that the consumption of Vioxx might be associated with an increased risk of cardiovascular disease. Under the sub‑heading “Interactions” in the “Precautions” section the following appeared:
Aspirin
Concomitant administration· of low‑dose aspirin with Vioxx may result in an increased rate of GI ulceration or other complications, compared to use of Vioxx alone. At steady state, Vioxx 50 mg once daily had no effect on the anti‑platelet activity of low‑dose (81 mg once daily) aspirin, as assessed by ex vivo platelet aggregation and serum TXB2 generation in clotting blood. Vioxx is not a substitute for aspirin for cardiovascular prophylaxis.
In the “Adverse Reactions” section, it was said that Vioxx had been evaluated for safety in approximately 5400 subjects/patients in blinded, controlled, clinical trials, at various doses ranging from 5 mg to 1000 mg. In general, it was said, treatment with Vioxx was well‑tolerated. With respect to clinical adverse experiences which had occurred in 2% or more of subjects/patients in the osteoarthritis trials, the only entry under the heading “Cardiovascular System” was “Hypertension”, where it was stated that 3.5% of those taking Vioxx had encountered a serious adverse experience. The corresponding number for placebo was 1.3%, for ibuprofen 3.0%, and for diclofenac 1.6%. In the same trials, there had been certain “spontaneous adverse events [occurring] in >0.1% to 1.9% of patients treated with Vioxx regardless of causality”, which included the following:
Cardiovascular System: angina pectoris, atrial fibrillation, bradycardia, haematoma, irregular heart beat, palpitation, premature ventricular contraction, tachycardia, venous insufficiency.
Then, with respect to experiences encountered by fewer than 0.1% of patients, regardless of causality, the Product Information identified the following:
Cardiovascular: cerebrovascular accident, congestive heart failure, deep venous thrombosis, myocardial infarction, pulmonary embolism, transient ischaemic attack, unstable angina.
Introduction of Vioxx to the Australian Market
226 It seems that the first formal indication to the medical profession that Vioxx was available for prescription for the symptomatic treatment of osteoarthritis was the sending of a “Dear Doctor” letter in October 2000. The letter said:
[MSDA] are pleased to advise the availability of VIOXX …
VIOXX is a COX-2 inhibitor and is indicated for the symptomatic treatment of osteoarthritis (OA).
VIOXX offers:–
Strength: VIOXX has the power of high doses of the conventional NSAID, diclofenac.
GI Safety: The incidence of serious GI complications (PUBs*) was significantly less with VIOXX than with the conventional NSAIDs ibuprofen and diclofenac.
Simplicity: Prescribing features of VIOXX include:
· use with a wide range of commonly prescribed drugs without evidence of adverse interactions†
· no sulfonamide contraindication
· no dosage adjustments are necessary for elderly patients, or patients with mild to moderate hepatic or renal insufficiency
· once‑daily dosage
* perforations, ulcers and bleeds
† Interactions with warfarin, methotextrate and lithium may occur. Usual monitoring should be followed. For details refer to enclosed Product Information.
VIOXX is available in 12.5mg and 25mg strengths in packs of 30 tablets.
At this stage VIOXX is not listed on the PBS so it will be available as a private prescription only. A rebate may be available for patients with private health insurance, from their health fund.
VIOXX represents a significant advance in the management of osteoarthritis. Full Product Information is included with this letter. Please review before prescribing. Should you have any queries regarding the product, please do not hesitate to call our Medical Information Group … .
The letter was signed by Ms Jane Paterson, Senior Product Manager. Her reference to “the PBS” was to the Pharmaceutical Benefits Scheme (“PBS”).
227 In the same month a “Dear Pharmacist” letter was sent out by MSDA. It announced the availability of Vioxx, “a once‑daily COX-2 inhibitor … indicated for the symptomatic treatment of osteoarthritis”. Doses and wholesale prices were referred to. It was said that Vioxx was not yet listed on the PBS. A copy of the Product Information was included with the letter.
228 Another “Dear Pharmacist” letter was sent in December 2000, and announced the listing of Vioxx on the PBS, taking effect from 1 February 2001. It was said that Vioxx was “a new once‑daily COX-2 inhibitor”. The letter continued:
Vioxx represents a significant advance in the management of osteoarthritis. Addressing the issue of GI safety associated with conventional NSAID therapy, VIOXX offers superior tolerability without compromising efficacy.
VIOXX inhibits COX-2 without having a clinical meaningful effect on COX-1.
The recommended starting dose is 12.5 mg once‑daily, however some patients may receive additional benefit by increasing the dose to 25 mg once-daily. The maximum recommended daily dose is 25 mg.
VIOXX is available from your wholesaler in two strengths, 12.5 mg and 25 mg in packs of 30 tablets.
Again, a copy of the Product Information was attached to the letter.
229 The listing of Vioxx on the PBS − which brought with it the government subsidy − coincided with the official “launch” of the Vioxx marketing campaign (to which I shall refer further below). Mr Back said (in his oral evidence) that Vioxx was not “marketed” before February 2001, by which he meant that it had not been launched and was not available to the general public. He accepted, however, that there was a period of time prior to the formal launch of Vioxx in which sales representatives were allowed to discuss the product with physicians, and that, at this time, the marketing division of MSDA was actively trying to encourage knowledge, understanding and appreciation of Vioxx amongst specialists, general practitioners and other health care professionals. I think it fair to infer that activities along these lines were being undertaken from about the time of the sending of Ms Paterson’s “Dear Doctor” letter in October 2000.
Amendment to the Vioxx Product Information
230 The Vioxx Product Information was amended 11 times between December 1999 and September 2004. The only amendment to which detailed reference needs to be made in the present context is that which resulted from the findings in the VIGOR trial. Mr Back was advised about the interim results in a teleconference conducted by Ms Simpson on about 24 March 2000. Although not stated by, or put to, Mr Back, I infer that this was the conference to which I have referred at para 97 above. That being so, Mr Back would have seen the slides distributed by Merck to international subsidiaries for the purpose of that conference (see paras 98‑102 above). I do not, therefore, accept his evidence that the first written information he received was in an email from Ms Simpson on 27 March 2000. Mr Back said that, after receiving that email, he contacted Dr Chipman at the TGA, and told him that the VIGOR study had met its primary endpoint of demonstrating a difference in gastrointestinal outcomes in favour of Vioxx, but that there had also been a difference in serious cardiovascular outcomes in favour of naproxen. Mr Back said that MSDA wanted to file an application to include the results of VIGOR in the current osteoarthritis Product Information. Dr Chipman said that he understood that it might take some time to put a full application together, given that the results were, at that stage, interim ones only. He said that MSDA should immediately consider a safety‑related notification if the serious adverse events were not yet in the existing Product Information, and that the TGA would be happy to include important safety information in the current Product Information, no matter what the patient group.
231 On 11 April 2000, MSDA submitted an interim analysis addressing the cardiovascular and gastrointestinal findings of VIGOR to the TGA. In its covering letter, it stated that a final, complete, data package would be available in July, at which time it intended to make a formal application for inclusion of the study results in the Product Information. Mr Back said (in his evidence) that the reference to July was a mistake on his part, as he thought that this was the date when the complete data package was to be available.
232 In his witness statement, Mr Back noted the publication of Bombardier (2000) in November 2000, but it was apparently not until 12 January 2001 that a submission was made to the TGA to add the VIGOR information to the Vioxx Product Information. He explained that this was a Category 1 application because the requested changes to the Product Information did not meet the TGA’s criteria for a safety‑related notification, in that they involved revisions to the “Clinical Trials” section and the inclusion of a revised gastrointestinal safety precaution statement which had the effect of broadening the potential patient population.
233 In the covering letter for the application of 12 January 2001, it was stated:
The prevalence of NSAID‑associated side effects, particularly upper gastrointestinal perforations, symptomatic ulcers and upper‑GI bleeds is well known and is a significant public health concern. The current PI contains details of a combined analysis of clinical upper [gastrointestinal] events (PUBS) from all Phase IIb/III studies. These studies with VIOXX, a selective COX-2 inhibitor, demonstrated improved gastrointestinal safety compared with non‑selective COX-l/COX-2 inhibitors.
The Company has completed a large, prospective, outcomes clinical trial (VIGOR), assessing the incidence of serious upper gastrointestinal adverse events during chronic treatment with VIOXX or naproxen, in patients at risk for developing serious [gastrointestinal] toxicity. The results of this study provide further evidence of the improved [gastrointestinal] safety of VIOXX compared to non‑selective NSAIDs. We request that details of this clinical trial be added to the Product Information.
The letter itself made no reference to cardiovascular events, which Mr Back justified by noting that the purpose of the application was to assess the gastrointestinal outcomes of the VIGOR trial. Amongst the attachments to the letter was the amended Product Information being sought by MSDA. The only change which might be thought to have anything to do with the cardiovascular results of the VIGOR trial were passages that pointed out that Vioxx was not a substitute for aspirin, because of the absence of antiplatelet effects.
234 Attached to the application of 12 January 2001 were two clinical expert reports, one on the gastrointestinal findings of VIGOR by Dr Reicin, and the other on the cardiovascular findings by Dr Eliav Barr, both of MRL. We are presently concerned only with Dr Barr’s report. That report appears to be based on the report provided by MRL to the FDA on 13 October 2000, mentioned in para 131 above. Dr Barr set out the same table of CVT adverse experiences as was there referred to. He said that therapy with naproxen was associated with a 58% reduction in the risk for the development of such experiences compared with Vioxx therapy. He referred to four analyses that had been undertaken by Merck.
235 First, Dr Barr referred to the distinction between those patients who were, and those who were not, aspirin‑indicated. He noted that 31% of the confirmed CVT events in VIGOR occurred in the 4% of patients who were aspirin‑indicated. In that group, naproxen therapy was associated with an 80% reduction in risk. In the non‑aspirin indicated group (96% of the patients) naproxen therapy was associated with a 43% reduction in risk. He continued:
The high rate of cardiovascular events in the “aspirin‑indicated” sub‑cohort is not surprising. Patients with symptomatic atherosclerotic cardiovascular disease are at particularly high risk of atherosclerotic plaque rupture that can result in arterial thrombosis and recurrent organ ischemia/infarction. Thus, if a study is evaluating compounds with differential effects on cardiovascular homeostasis, such differences would most likely be observed in a high risk cohort such as this group.
Secondly, noting that patients who received Vioxx had a higher incidence of hypertension and oedema‑related adverse experiences compared with patients who received naproxen, Dr Barr asked himself whether that circumstance had caused the imbalance in cardiovascular events in the study. He concluded that it had not. Thirdly, he analysed the VIGOR results by reference to the APTC endpoint, and found that risk was reduced by 49% in the naproxen group relative to the Vioxx group. He noted that the difference in the event rates between the treatment groups was due primarily to a difference in the rates of myocardial infarction, and that the incidence of thrombotic strokes was similar between treatment groups. Fourthly, he looked for evidence, other than the CVT results as such, of the antiplatelet effect of naproxen in the VIGOR trial, such as bleeding. He found that, compared with Vioxx, naproxen was associated with a 2.1‑fold and 3.5‑fold higher incidence of ecchymosis and epistaxis, respectively. He continued:
These results also suggest that naproxen therapy was accompanied by high‑level platelet inhibition in the VIGOR study. Such inhibition may have contributed to the observed difference in the rates of thrombotic cardiovascular serious adverse experiences between the naproxen and rofecoxib groups in the study. However, it is also fair to say that the relationship between such in vivo manifestations of medication‑induced platelet inhibition and the effect of such medications on cardiovascular event rates has not been well defined.
236 Dr Barr said that there were three potential hypotheses to explain the cardiovascular findings of the VIGOR study, namely: (1) Vioxx caused a prothrombotic or otherwise deleterious perturbation of cardiovascular homeostasis; (2) naproxen had potent antiplatelet properties throughout its dosing interval; and (3) the findings were a play of chance, given the relatively small number of cardiovascular events in the study. He said that, taking the VIGOR results in isolation, it was difficult to eliminate any of these hypotheses from consideration.
237 Considering whether Vioxx was prothrombotic, Dr Barr referred to the results of other studies involving the drug, to the extent that they were then available (his data was current at 15 September 2000). He commenced with the Alzheimer’s disease studies. His conclusion with respect to them was:
[T]here was no evidence in these studies that therapy with rofecoxib was accompanied by increased cardiovascular risk compared with placebo in this elderly, relatively high‑risk patient population. These results substantially reduce the likelihood that the cardiovascular findings in the VIGOR study were due to rofecoxib‑mediated prothrombotic or otherwise deleterious perturbations of cardiovascular homeostasis.
238 Dr Barr turned next to the Phase IIb/III osteoarthritis studies. He noted that they involved comparisons of Vioxx with NSAIDs, although in the case of some studies there were also placebo arms. He said that, in the NSAID comparison, the incidence of CVT events was 2.05 events per 100 patient‑years in the Vioxx arms compared with 2.27 events per 100 patient‑years in the NSAID arms. When the comparator was placebo, the figures were 2.48 and 2.36 respectively, although Dr Barr said that the number of events was small. His conclusion was:
Thus, in this group of studies, therapy with rofecoxib and the nonselective NSAIDs (mostly diclofenac and ibuprofen) was associated with comparable incidences of thrombotic cardiovascular events.
In the group of placebo‑controlled studies, there did not appear to be any imbalances in cardiovascular event rates between treatment groups. However, there were too few patient‑years of follow‑up and thrombotic cardiovascular events in these trials to draw firm conclusions regarding the relative risk for the development of such events associated with rofecoxib relative to placebo in these studies.
239 Dr Barr next set out the results of the ADVANTAGE study, a gastrointestinal outcomes study conducted between March 1999 and April 2000, of 12 weeks’ duration for each patient, comparing Vioxx with naproxen. He noted that there were too few events recorded in that study to enable any conclusions to be drawn. He expressed effectively the same view about the results of Merck’s studies of Vioxx in patients with rheumatoid arthritis (other than VIGOR). Finally in this section, Dr Barr mentioned MRL’s Worldwide Adverse Experiences Surveillance database, in which it was recorded that there were, as at 13 March 2000, 78 reports of CVT events, a rate of 8.1 reports per 100,000 patient‑treatment-years distributed. He said that this rate was low relative to the background occurrence of such events in the age group of patients in which Vioxx was prescribed, and was not disproportionate to the numbers reported with two other medications marketed by Merck that did not have any known associated cardiovascular risks and which were indicated for treatment populations of similar age (finasteride and alendronate).
240 The next section of Dr Barr’s report was headed “Overall Assessment of Cardiovascular Outcomes in Clinical Studies of Rofecoxib and Suggested Guidance to Physicians and Patients”, and commenced with an “Overview”. Dr Barr said:
Platelet aggregation is central to the development of thrombotic cardiovascular events such as [myocardial infarction] … Agents that inhibit COX‑1, such as aspirin, substantially inhibit platelet aggregation and prolong bleeding time … Aspirin and other compounds that generate nearly complete inhibition of platelet aggregation have been shown to reduce the incidence of thrombotic cardiovascular events in high‑risk patients … Because COX‑2 selective inhibitors do not block COX‑1‑mediated platelet aggregation, it was hypothesized that therapy with nonselective COX‑1/COX‑2 inhibitors may result in a lower incidence of thrombotic cardiovascular events in high-risk patients compared with COX‑2 selective inhibitors. Furthermore, the recent demonstration that COX‑2 inhibitors (rofecoxib and celecoxib) selectively suppress the synthesis of prostacyclin (which has vasodilatory and antiplatelet properties) without inhibiting the synthesis of thromboxane (which has vasoconstrictive and proaggregatory properties) has fuelled speculation that these compounds may act as prothrombotic agents …
After referring again to the mixed messages that Merck had received from its various studies, Dr Barr said that it was important to address the following questions:
1) Is there evidence to suggest that chronic therapy with naproxen, a nonselective COX-l/COX-2 inhibitor may result in improved cardiovascular outcomes compared with therapy with COX‑2 selective inhibitors such as rofecoxib? If the answer is yes, is this protective effect unique to naproxen, or consistent across the entire class of nonselective COX‑1/COX‑2 inhibitors?
2) Is there evidence to suggest that the cardiovascular findings of the VIGOR study were due to a play of chance?
3) Is there evidence to suggest that chronic therapy with COX‑2 selective inhibitors such as rofecoxib may result in an increased cardiovascular risk?
4) Based on (1) to (3), what guidance should be given to physicians and patients regarding the optimal use of COX‑2 selective inhibitors such as rofecoxib?
241 Dr Barr then turned to an explanation of the mechanism of non‑selective COX-1/COX-2 inhibitors, commencing with aspirin. He explained its antiplatelet action. He next dealt with the like action of other NSAIDs, observing that, if they were to act as “vascular‑protective” agents, “near complete inhibition of TXA2 sustained over time is needed”. He referred to Protocol 061 in some detail, and concluded:
Thus, this study demonstrated a gradient of antiplatelet effects among the nonselective NSAIDs and the COX-2 selective inhibitor rofecoxib: therapy with naproxen was associated with antiplatelet effects similar to those reported for aspirin (that is, near‑maximal inhibition of platelet aggregation sustained throughout its dosing interval); therapy with diclofenac and ibuprofen resulted in modest antiplatelet effects over their respective dosing intervals; and therapy with rofecoxib (and placebo) was not associated with substantive antiplatelet effects.
242 Dr Barr referred to other such studies in which similar results had been observed, and drew a distinction between NSAIDs (such as, in his view, naproxen) which had sustained, near‑maximal, COX-1-inhibiting properties over the dosing interval and NSAIDs which did not. Of the former, he said that they “may very well provide a vascular protection efficacy profile similar to that of aspirin”. In the course of that discussion, Dr Barr referred to the two studies which generated the FitzGerald hypothesis, and pointed out that therapy with rofecoxib had been shown to reduce the production of the urinary metabolite of prostacyclin by 61%. He continued:
It has been proposed that the observed reduction in PGI2 synthesis (without corresponding reductions in TXA2 synthesis) associated with COX-2 selective inhibitors could theoretically lead to a mild proaggregatory state in patients receiving these compounds. However, a reduction in PGI2 synthesis of 60% may not be physiologically meaningful given the observation that there were no observed increases in the incidence of thrombotic cardiovascular events in patients who received rofecoxib relative to placebo in the Protocols 078 and 091 (the placebo‑controlled studies of chronic rofecoxib therapy in Alzheimer’s disease patients).
243 Under the heading “Cardiovascular Clinical Studies of Reversible Nonselective COX-l/COX-2 Inhibitors”, Dr Barr referred to two studies which, despite confusing footnotes, appear to have been Brochier (1993) and Fornaro (1993). Again, he proposed that the data from them “suggest that nonselective COX-l/COX-2 inhibitors that generate potent, sustained COX-l inhibition have vascular‑protective properties similar to those observed with aspirin.”
244 After summarising his views as to the cardioprotective effect of naproxen, Dr Barr turned to an“Assessment of the Cardiovascular Effects of Therapy With COX-2 Selective Inhibitors Such as Rofecoxib”. Under that heading, he articulated, and argued the case in favour of, the following propositions:
· Naproxen mediates aspirin‑like antiplatelet effects. Rofecoxib does not mediate antiplatelet effects. Thus, the reduction in the incidence of thrombotic cardiovascular serious adverse experiences in patients who received naproxen compared with patients who received rofecoxib in the VIGOR study is not surprising.
· Exclusion of concomitant use of aspirin or antiplatelet agents, and the enrollment of a subpopulation of patients in whom aspirin therapy was indicated, created a study subpopulation at high risk for thrombotic cardiovascular events.
· Protocol features that reinforced compliance with the protocol‑specified study medication regimen ensured a rigorous comparison of COX-2 selective inhibition and sustained, potent nonselective COX-1/COX-2 inhibition.
· Selection of a rheumatoid arthritis patient population for evaluation in the VIGOR study was logical from a GI perspective, but resulted in the selection of a patient cohort whose underlying disease confers increased cardiovascular risk.
Dr Barr’s conclusion on this aspect was:
Overall, there is strong evidence to suggest that naproxen’s aspirin‑like antiplatelet effects on one hand and the absence of such effects following rofecoxib therapy were responsible for the cardiovascular findings of the VIGOR study.
245 Dr Barr next considered whether the cardioprotective effect to which he referred was unique to naproxen, or was shared by all non‑selective NSAIDs. He concluded that there was “strong evidence” that the effect was seen only in a subclass of such NSAIDs. He then considered whether the VIGOR results might be explained as a “play of chance”, and concluded that they should not. In this respect, he said:
Although the VIGOR protocol did not include a hypothesis to address cardiovascular findings or standardized methods for assessment of cardiovascular events, MRL had formulated a general exploratory hypothesis that therapy with nonspecific COX-l/COX-2 inhibitors and COX-2 selective inhibitors such as rofecoxib might result in differential vascular‑protective effects. This hypothesis was based on the differential effects of nonspecific COX-l/COX-2 inhibitors and the COX-2 selective inhibitors celecoxib and rofecoxib on thromboxane and prostacyclin metabolism observed in clinical pharmacology studies. Also, the Adjudication SOP was a rigorous process that efficiently identified and classified thrombotic cardiovascular events. Finally, as described above, the findings in the VIGOR study are biologically plausible, given the known benefit of aspirin/antiplatelet therapy in patients at risk for the development of thrombotic cardiovascular events and the differential antiplatelet effects of naproxen and rofecoxib demonstrated in clinical pharmacology studies (and indirectly in the VIGOR study).
Overall, it is likely that the cardiovascular results of the VIGOR study represent a true difference in cardiovascular outcomes between chronic naproxen and rofecoxib therapy in a cohort of rheumatoid arthritis patients that included a subgroup of patients with an indication for chronic vascular‑protective aspirin therapy in whom such therapy was not given.
246 Dr Barr then asked the question: “Is there evidence to suggest that chronic therapy with COX-2 selective inhibitors such as rofecoxib results in an increased cardiovascular risk (i.e. compared to placebo)?” The whole of his reasoning in this respect was as follows:
Although COX-2 selective inhibitors such as celecoxib and rofecoxib could theoretically generate a mild pro‑aggregatory state in some patients due to partial inhibition of PGI2 synthesis, I do not believe that it is likely that this effect contributed to the cardiovascular results of the VIGOR study. This conclusion is supported by the following observations:
1) In clinical trials that compared rofecoxib to placebo (studies in early Alzheimer’s disease, a subset of studies in osteoarthritis patients), there were no differences in the incidences of thrombotic cardiovascular serious adverse experiences between treatment groups. These studies evaluated patients with the greatest baseline cardiovascular risk of all the patients evaluated in the rofecoxib clinical program. Thus, the absence of an imbalance in the incidence of thrombotic cardiovascular events in these studies argues strongly against a prothrombotic or otherwise deleterious effect of rofecoxib on cardiovascular homeostasis.
2) In clinical trials in osteoarthritis patients that compared rofecoxib with diclofenac and ibuprofen, a nonspecific COX-l/COX-2 inhibitor without sustained, near‑complete inhibition of platelet aggregation, there were no differences in the incidences of thrombotic cardiovascular serious adverse experiences between treatment groups. Again, the absence of an imbalance in cardiovascular event rates between rofecoxib and its comparators is reassuring.
3) The absolute rates of serious thrombotic cardiovascular serious adverse experiences were similar in patients treated with rofecoxib in both the Phase IIb/III osteoarthritis combined analyses and VIGOR, suggesting that the difference between rofecoxib and naproxen in VIGOR was driven by a reduction in events in the naproxen group rather than an increase in event rates in the rofecoxib arm.
Dr Barr went on to conclude that there was “no evidence to suggest that therapy with rofecoxib is prothrombotic”, because CVT event rates were comparable in trials that compared rofecoxib to placebo or to non-selective NSAIDs with modest antiplatelet properties, and the absolute CVT event rates observed in the VIGOR study were comparable to those observed in the Phase III osteoarthritis program. He recommended that the Product Information contain “general guidance” to physicians and other health professionals which reflected that conclusion.
247 The next thing that happened was the making, on 3 July 2001, of a clinical evaluation report within the TGA. The clinical evaluator noted that MSDA had sought amendments to the Vioxx Product Information in the following areas:
· the addition of an introductory statement under the heading “gastrointestinal safety”;
· the inclusion of a description of the VlGOR study in the “Clinical trials” section and the inclusion of the results;
· modification of the “precautions section”;
· modification of the “adverse reactions section”.
248 Of Dr Barr’s report, the clinical evaluator said:
The review of the cardiovascular effects was particularly helpful. The author pointed out that the effects of rofecoxib on cardiovascular events was [sic] likely to be real, and possibly due to the lack of platelet inhibition. He suggested that the patient population in the VIGOR study was more at risk of cardiovascular events and was not treated with prophylactic aspirin, and this combination of factors lead to the excess of cardiovascular events in the rofecoxib group. I do not agree with his assessment of the likelihood of increased risk as it is a rheumatoid arthritis population rather than osteoarthritis (ie not as old). However, the expert does include some usefu1 recommendations about the potential need to treat at risk patients with low dose aspirin as well as rofecoxib; unfortunately, there are no data in this dossier to support the safety of this combination.
The clinical evaluator noted that –
… the adverse reactions section that is proposed makes no mention of the cardiovascular event data, which in my opinion, is completely unacceptable, particularly given the comments of Merck’s own expert report!
He or she recommended that the cardiovascular results of VIGOR should be set out in more detail than apparently MSDA had proposed, with a cross‑reference to the “Adverse effects” section. In the “Precautions” section, it was proposed that there be a new paragraph in the following terms:
In the VlGOR study, treatment with rofecoxib was associated with a higher incidence of cardiovascular events than treatment with a non‑selective COX-1/COX-2 inhibitor, naproxen. These events included hypertension, the development of peripheral oedema, cardiac failure and myocardial infarction. It is possible that this difference was due to lack of effect on platelet aggregation in a population that is potential [sic] at increased risk of cardiovascular events (although the population in the VIGOR study was not generally at increased risk.) It is recommended that patients who are taking rofecoxib for extended periods should be assessed for cardiovascular risk and consideration may need to be given to the concomitant use of low dose aspirin. However, the safety of this combination of treatments has not been established, and physicians should assess the likely benefits and risks of long term rofecoxib therapy carefully.
249 The clinical evaluator expressed his or her conclusions as follows:
The VIGOR study contains important data that should be included in the product information document. It obviously is insufficient to establish efficacy for the indication ‘rheumatoid arthritis’ but this is not a major concern given that the major focus of the study is a question of comparative safety. Provided the changes to the draft PI that are listed above are included, the application could be approved.
The issue of concern that is not adequately resolved by the study and that may need to be the subject of further consideration by ADRAC is the question of the serious cardiovascular events. On the basis of these data, it is clear that the company should be asked to comment on the overall risk/benefit of rofecoxib, and that there needs to be ongoing monitoring of the cardiovascular effects of this drug.
Mr Back explained that “ADRAC” is the Adverse Drug Reactions Advisory Committee, a subcommittee of ADEC. He accepted that the marketing staff within MSDA would have been aware of the doubts expressed by the evaluator.
250 On 2 August 2001, the TGA wrote to MSDA enclosing a copy of the summary and “proposed action” prepared by the delegate who was to decide the outcome of MSDA’s application of 12 January 2001. The delegate, Dr Christine Anantharajah, referred to Dr Barr’s report and said that the osteoarthritis, Alzheimer’s disease and rheumatoid arthritis studies on which he had relied had not been evaluated or were then under evaluation and continued:
Hence, [Dr Barr’s report] is of minimum benefit to this application.
The claim that the excess cardiovascular events in the rofecoxib group could be explained by the cardioprotective effect (due to the antiplatelet properties) of naproxen is not supported, as,
1. There are no prospective placebo controlled studies of naproxen that show that transient inhibition of platelet aggregation is effective in decreasing the risk of cardiovascular events.
2. The effect size of naproxen in reducing CV thrombotic events relative to rofecoxib in the VIGOR study (58%) exceeds the effect size of aspirin compared to placebo in other trials (25% to 30%) …
Having noted the recommendation of the clinical evaluator, Dr Anantharajah provided her own comments, which included the following:
4. The findings relating to cardiovascular adverse events need to be included in the PRECAUTIONS section of the PI. Low dose aspirin is recommended for those taking rofecoxib over extended period of time to address the increase in thrombotic effects. This is supported by the evaluator. However, I do not recommend this as there are no data to support this claim. The sponsor should generate long term data on low dose aspirin with rofecoxib prior to making suggestions in the PI about concomitant therapy.
5. There should be a statement that long term therapy is not recommended for those with a history of cardiovascular disease, especially, as the VIGOR study excluded those with cardiovascular disease and those requiring low dose aspirin for cardioprotection. The excess reports of cardiovascular thrombotic events in the rofecoxib treated group also supports this statement.
251 Mr Back accepted that the delegate disagreed with MSDA that the most likely explanation for the VIGOR cardiovascular results was the beneficial effect of naproxen. He said that no‑one in MSDA had undertaken any investigation as to whether the naproxen effect was greater than the corresponding aspirin effect; and nothing was put to the TGA to demonstrate that naproxen had anti‑thrombotic effects substantially greater than aspirin.
252 On 10 September 2001, MSDA submitted to the TGA its “Pre‑ADEC Response”, in which it commented on the reports of the Clinical Evaluator and Dr Anantharajah. The response as such reiterated MSDA’s interpretation of the VIGOR results, in the light of the other data which had been referred to by Dr Barr. However, MSDA accepted the recommendation that a new passage of text about the cardiovascular results of the VIGOR trial be inserted into the “Precautions” section of the Product Information. It proposed the following:
Cardiovascular Effects
A large clinical trial in rheumatoid arthritis patients has compared the long‑term safety of rofecoxib 50 mg once daily (twice the maximum recommended dose) and naproxen 500 mg twice daily. The rate of serious cardiovascular thrombo‑embolic adverse events was significantly lower in patients receiving naproxen than in the rofecoxib group: 0.70 events per 100 patient‑years compared with 1.67 events per 100 patient‑years. In other controlled clinical trials, spontaneous reports of these cardiovascular events were similar between VlOXX and non‑selective NSAID comparators. The difference in antiplatelet activity between some COX-1 inhibiting NSAlDs and COX-2 selective inhibitors may be of clinical significance in patients at risk of thrombo‑embolic events.
253 At its meeting on 4‑5 October 2001, ADEC resolved that there should be no objections to the inclusion of the VIGOR findings in the Product Information for Vioxx, subject to the finalisation of the terms thereof, which should include a statement to the effect that long‑term therapy was not recommended for those with a history of cardiovascular disease, and should not include a recommendation that low‑dose aspirin be used by patients taking Vioxx over an extended period of time.
254 On 15 October 2001, the TGA made a s 31 request to MSDA which, relevantly for present purposes, required the amendment of the draft Product Information submitted on 10 September 2001 by the inclusion of a statement that “long term therapy is not recommended for patients with a history of cardiovascular disease”.
255 On 17 October 2001, Mr Back faxed a copy of the ADEC resolution and the s 31 request to Ms Bea Loren, the labelling scientist at Merck, whose role was to co‑ordinate the scientists in the Merck group in developing the worldwide physician circular, the group’s core data documents and each of the individual country’s Product Information documents. In a contemporaneous email, Mr Back drew attention to the respects in which ADEC had required changes in the Product Information as submitted by MSDA. In a reply of 19 October 2001, Ms Loren said:
Well, I think, and as you suggest, we should keep the concom aspirin use, given that this is in the Interaction section and is not intended to be a recommendation. It’s a clinpharm statement. The CV sentence is probably more difficult. Do you think we have a chance to argue against the inclusion of such wording? Especially the non recommendation for “long term therapy” does not seem to have a justification on the basis of the data. And if we eliminate this part, what’s left is already said in the previous sentence (more or less). How long can we negotiate the label? Shall we have a teleconf to outline our strategy?
In a response to Ms Loren of the same date, Mr Back said that he did not think that MSDA would be able to argue against the inclusion of the recommendation against long term therapy, adding that both the clinical evaluator and the delegate, as well as ADEC itself, had requested it. He concluded: “We should try to play with the wording to make it more palatable.” Mr Back denied that he meant “palatable” in a marketing sense, referring to Ms Loren’s objective to standardise labelling across the world.
256 On 2 November 2001, MSDA responded to that request, but proposed only that the following be added to the draft of 10 September: “Physicians should assess the importance of these data for an individual patient when prescribing long term therapy with a COX-2 inhibitor”. In the letter in support of that proposal, MSDA said:
At present, the cardiovascular findings in the VIGOR stand without an established explanation. The Company believes that the preponderance of available data strongly suggest the imbalance in thromboembolic events stemmed from cardioprotective properties of naproxen, lacking with rofecoxib. Plausibility comes from known, sustained, inhibitory effects of naproxen, in comparison with many other nonselective NSAIDs, on platelet aggregation, and the VIOXX meta‑analysis data showing no increased risk of thromboembolic cardiovascular events for rofecoxib, compared with placebo or a non‑naproxen NSAID group of comparators (ibuprofen diclofenac, and nabumetone, none of which imparts sustained inhibitory effects on platelets).
Mr Back confirmed that this was MSDA’s view at the time. However, he also accepted that there was nothing that he had been given which would suggest that any cardioprotective effect of naproxen was substantial enough to explain the VIGOR results. He accepted that, within MSDA at the time, while it was considered that the cardioprotective work of naproxen was the most likely explanation for the VIGOR results, there was nonetheless a view that a pro‑thrombotic tendency of Vioxx was a plausible alternative explanation. In the course of cross‑examination, it was put to him that, by advancing only the naproxen explanation, MSDA was misleading the TGA. He rejected that. He pointed out that the delegate had made it quite clear that she understood the pro‑thrombotic explanation, and that MSDA was in effect countering that with its own view of the science.
257 It appears that, at about this time, a new delegate was appointed in place of Dr Anantharajah. It was Dr Andrew Pengilley. He telephoned Mr Back on 6 November 2001, and sought a revision of the proposed text for the PI by the inclusion of the words “at risk for cardiovascular disease”. Mr Back informed Ms Loren of this wording, and her reaction was that the expression “cardiovascular disease” was far too broad. The following day, Mr Back responded to Dr Pengilley with the following proposal: “Physicians should assess the importance of these data for an individual patient at risk for cardiovascular thrombo‑embolic events, if considering long term therapy with a cox-2 selective inhibitor.” Dr Pengilley called again the same day to inform Mr Back that the Product Information had been approved. Formal approval was given on 16 November 2001.
258 In the “Precautions” section of the Product Information as approved on 16 November 2001, a new sub‑heading was introduced, “Aspirin”, under which it was stated that Vioxx was not a substitute for aspirin for cardiovascular prophylaxis because of its lack of effect on platelets. Then another new sub‑heading, with its corresponding text, was introduced as follows:
Cardiovascular Effects
A large clinical trial in rheumatoid arthritis patients has compared the long‑term safety of rofecoxib 50 mg once daily (twice the maximum recommended dose) and naproxen 500 mg twice daily. The rate of serious cardiovascular thrombo‑embolic adverse events was significantly lower in patients receiving naproxen than in the rofecoxib group: 0.70 events per 100 patient‑years compared with 1.67 events per 100 patient‑years. In other controlled clinical trials, spontaneous reports of these cardiovascular events were similar between VIOXX and nonselective NSAID comparators. The difference in anti platelet activity between some COX-l inhibiting NSAIDs and COX-2 selective inhibitors may be of clinical significance in patients at risk of thrombo‑embolic events. Physicians should assess the importance of these data for an individual patient at risk for cardiovascular thrombo‑embolic events, if considering long term therapy with a Cox-2 selective inhibitor.
From then until the withdrawal of Vioxx in September 2004, there were further amendments made to the Vioxx Product Information, but none that require mention in the present context.
The Marketing of Vioxx in Australia
259 The applicant led a deal of evidence as to the means by which Vioxx was marketed in Australia by MSDA. For the most part, this was done by the tender of documents discovered by MSDA. Many of those documents were provided by the applicant to one of his expert witnesses, Prof Robert Donovan, Professor of Behavioural Research at the Centre for Behavioural Research in Cancer Control, Division of Health Sciences, Curtin University; Director, Mentally Healthy WA, Division of Health Sciences, Curtin University; Co-Director, Social Marketing Research Unit, School of Marketing, Curtin University; and Professor of Social Marketing in the School of Marketing, Curtin Business School, Curtin University. Prof Donovan referred to a number of those documents in his report. For reasons explained at paras 855‑858 below, I found both the documents and the evidence of Prof Donovan of limited utility. In this part of my reasons, I shall undertake a purely factual survey of the main classes of documents which related to the marketing efforts of MSDA, and to the means employed generally to promote Vioxx. It is not intended to be an exhaustive survey: such an undertaking would burden these reasons with much unhelpful material. It is, however, intended to draw attention to the documents which, for reasons which will become clear, are relevant on any view, and to provide a factual context against which the applicant’s marketing case may be assessed.
260 The evidence establishes that MSDA used a variety of means of communication to promote awareness of, and ultimately the use of, Vioxx. However, the respondents did not themselves lead evidence of any communication − written or oral − made individually to any doctor, pharmacist or health care professional. They called two witnesses in response to the applicant’s marketing case. The first was Ms Penelope Dobson. Between March 1999 and June 2001, she was one of two Sales & Marketing Directors employed by MSDA. In June 2001, as a result a restructure within MSDA, the sales and marketing functions were split. The marketing department was divided into a number of “franchises”, to each of which were allocated certain products. Ms Dobson became Marketing Director for the “Musculoskeletal Franchise”, which was responsible for the marketing of Vioxx. She held this role until January 2003, after which she was not involved in the day to day operation of MSDA’s marketing division. The other witness was Mr Keiran McAuley, Senior Sales Manager (Hospital) for the National Specialty Division at MSDA. In early 2001 when Vioxx was launched, he was a representative in the “Musculoskeletal Team”, promoting prescription medicines to general practitioners in the Mornington Peninsula and surrounding areas in Victoria. On 1 April 2001, he was promoted to the position of Sales Manager Primary Care for Victoria and Tasmania, which new position gave him responsibility for the Musculoskeletal Team for the whole of Victoria. The evidence of Ms Dobson and of Mr McAuley was concerned with the practices, procedures and protocols followed by the marketing and sales departments of MSDA. To that extent, it was illuminating and useful, in ways to which I shall refer. It did not, however, descend to the details of communications actually made to a third party in relation to Vioxx.
261 That is not to say that there was no such evidence. It seems that MSDA discovered a great many documents that generally bespoke the existence of such communications, and in some cases I am able to infer that communications in corresponding terms were in fact made. However, as will become apparent, in at least one important respect I have not been satisfied that a generic pro‑forma document discovered by MSDA was in fact sent as apparently intended.
262 Doubtless reflecting the position of Vioxx as a prescription‑only medicine, the focus of MSDA’s marketing activities, at least to the extent revealed in the evidence, was upon medical practitioners. As I shall mention below, there were also some communications with pharmacists. There was no evidence of promotional activities directed towards other health care professionals. Save for one media release (the release or publication of which was not proved) there was no evidence of such activities directed towards the public generally in Australia. Clearly the interface with doctors was the most important area for the promotion of Vioxx, and both sides conducted their cases in court by reference to that assumption.
263 The most direct means by which MSDA communicated with medical practitioners generally was the transmission from time to time of pro‑forma “Dear Doctor” letters. There were five such letters in evidence. Because they are few in number, I shall refer to the terms of each of them. There is also a question as to which of these letters was in fact sent to practitioners. All five were tendered by the applicant, but not always for the same purpose. Clearly the applicant was in no position to give evidence that a particular letter had been sent, and none of the three general practitioners called gave evidence that he had received one. In the case of four of the letters, the respondents submitted that they were “final approved pieces” which were “in fact made available to the prescribing community”. I take this to be a submission that the documents in question were not merely drafts or versions in the course of preparation, but had official approval to be sent, and were in fact sent, to medical practitioners.
264 The first “Dear Doctor” letter in evidence was that to which I have referred in para 226 above. It was sent in October 2000. Ms Dobson described it, not inaptly in my view, as an “announcement letter”. The respondents included this letter on their list of “final approved pieces”. The letter was tendered by the applicant through Ms Dobson (in the course of cross‑examination). It was effectively common ground that the letter was sent to medical practitioners generally.
265 In August 2001, Mukherjee (2001) was published in the Journal of the American Medical Association. In evidence are two similar letters, each from MSDA and using the greeting “Dear Healthcare Provider”, dated 28 August and 3 October 2001. The letters took issue with the scientific basis of the article with respect to VIGOR. The first letter stated:
The reported differences in the incidence of myocardial infarctions (MI) between Vioxx and naproxen in the … VIGOR trial …, which is the main focus of the JAMA paper, can be explained by naproxen’s aspirin‑like ability to inhibit platelet aggregation and reduce cardiovascular events. Unlike some NSAIDs, Vioxx does not inhibit platelet aggregation (a desirable attribute when trying to avoid gastrointestinal adverse events) and would not be expected to reduce cardiovascular events as has been shown with naproxen in the VIGOR trial.
The corresponding paragraph in the second letter stated:
The main focus of the JAMA article relates to the reported difference in the incidence of myocardial infarction (MI) between Vioxx and naproxen in the … VIGOR trial … An explanation for this difference is based on naproxen’s ability to inhibit thromboxane A2 synthesis and platelet aggregation. As a result of this activity, naproxen may reduce the incidence of cardiovascular events in a manner similar to that observed with aspirin. Unlike some NSAIDs, Vioxx does not inhibit platelet aggregation (a desirable attribute when trying to avoid gastrointestinal adverse events) and would not be expected to reduce cardiovascular events.
Towards the end of both letters, it was stated:
Patient safety is of paramount importance to MSD. We routinely review data from completed studies and clinical use of our products, and consistent with this approach, we will continue to evaluate such data on agents that selectively inhibit COX-2 to enhance our understanding of these medicines and assess the potential value of future trials.
These letters were tendered without any comment of substance by the applicant, and without objection by the respondents. They were both on the respondents’ list of “final approved pieces”. It was, I consider, common ground that they were sent to medical practitioners generally.
266 The next “Dear Doctor” letter in evidence bore the date December 2001. It corresponded with the then recent amendment of the Vioxx Product Information to refer to the results of the VIGOR trial. It was unsigned, although over the name of the Medical Director of MSDA. It was in the following terms:
Merck Sharp & Dohme wishes to advise that the Product Information for VIOXX (rofecoxib, MSD) has been updated and includes new information of current interest.
– Results of VIGOR (VIOXX Gastrointestinal Outcomes Research) Trial – landmark study in over 8000 rheumatoid arthritis (RA) patients
The results from the VIGOR study confirmed the GI outcomes from previous studies and demonstrated a significant reduction in serious GI complications for patients treated with VIOXX compared to naproxen.
The results from VIGOR also raised the question as to whether COX-2 selective inhibitors increase the thrombotic risk of patients with RA. A recent meta‑analysis, which included a number of studies in RA patients, showed that except in the comparison to naproxen (in VIGOR), VIOXX was not associated with an excess of thrombotic events compared with either placebo or traditional NSAIDs (ibuprofen, diclofenac or nabumetone). Physicians should assess all patients at risk of thromboembolic events if considering long‑term therapy with COX-2 selective inhibitors.
– Low-dose aspirin for cardiovascular prophylaxis
Available evidence suggests that aspirin is underutilised in certain patients at risk of a cardiovascular event. To help ensure the appropriate prescribing of VIOXX and low‑dose aspirin, it is important to be aware of the following information:
– VIOXX can be used with low‑dose aspirin;
– Irrespective of traditional NSAID or COX-2 selective inhibitor use, low‑dose aspirin for cardiovascular prophylaxis should be considered for patients at risk of a thromboembolic event;
– VIOXX is not a substitute for aspirin for cardiovascular prophylaxis.
Before prescribing, please consult the accompanying full Product Information.
If you require any additional information, please call our Medical Information toll‑free number ….
267 The letter of December 2001 was tendered by the applicant as part of a large bundle of documents which were the subject of comment by Prof Donovan. The respondents did not object to it, but it was not one of their “final approved pieces”. Rather, they made the following submission about this and many other documents:
(a) unless otherwise specified, documents are properly characterised on the basis that they are not a promotional piece nor evidence of the information about Vioxx that MSDA, in fact, made available to the prescribing community;
(b) they do not evidence any departure by MSDA representatives from the [final approved pieces]; and
(c) in that regard, it is not sufficient for the applicant to attempt to side‑step the direct marketing material in favour of selective reference to draft, internal or non‑Australian documents.
This I take to be a submission that it had not been established on the evidence that the letter of December 2001 was in fact sent to medical practitioners. For his part, the applicant did not submit that the letter had been sent. There was no evidence (eg from a practitioner) that it had been received. As I have said (alone of the five letters here being dealt with), it was not signed. In the circumstances, I am not prepared to find that it was sent to medical practitioners generally.
268 The fifth and final letter in this sequence was dated March 2002, and had the purpose of announcing that Vioxx had been approved by the TGA for the symptomatic treatment of rheumatoid arthritis. Amongst other things, the letter stated:
In two efficacy studies involving approximately 2,000 patients with RA, Vioxx demonstrated a significant reduction in the number of tender/painful joints and number of swollen joints versus placebo. Vioxx 25 mg and naproxen 500 mg bd showed generally similar efficacy.
The landmark VIGOR Study conducted in 8,000 patients with RA showed that serious [gastrointestinal] complications (perforations, obstructions, ulcers and bleeds) were halved with Vioxx, 50 mg od (twice the recommended dose in RA) versus the comparator NSAID, naproxen, 500 mg bd.
This letter was also part of the bundle which related to Prof Donovan’s evidence, but it was not referred to by him. Rather, it was tendered by the applicant on the basis that it had been provided to him. There was no objection to the tender by the respondents, who accepted that it is what it purported to be. It was on the respondents’ list of “final approved pieces”. In the circumstances, I am prepared to find that it was sent to medical practitioners generally.
269 There were also five standard form “Dear Pharmacist” letters in evidence. Each was tendered by the applicant and described by the respondents as a “final approved piece”. Consistently with the way I have approached the “Dear Doctor” letters, therefore, I infer that each of these letters was sent to pharmacists generally. I have referred to the first two at paras 227 and 228 above.
270 The third “Dear Pharmacist” letter was dated August 2002, and reminded recipients that Vioxx was available as an oral suspension and in tablet form, at doses of 12.5 mg and 25.0 mg daily. The letter continued:
VIOXX is indicated for the symptomatic treatment of osteoarthritis and rheumatoid arthritis. Before dispensing VIOXX Oral Suspension, please review the enclosed product information.
We are aware that Naprosyn … suspension is being discontinued this year. As some of these patients may be switched to VIOXX Oral Suspension, you may like to ensure that your dispensary is stocked with a sufficient supply of this product. Merck Sharp & Dohme is adequately stocked with VIOXX Oral Suspension in order to meet the expected increase in demand. To order VIOXX Oral Suspension, please contact your pharmacy wholesaler.
If you would like any additional information about VIOXX, please contact your representative.
271 The fourth “Dear Pharmacist” letter was not dated, but, from the MSDA code endorsed on it, I infer that it too was sent around August 2002. Enclosed with the letter was an “arthritis information kit”, which included a copy of the Vioxx Product Information and Consumer Medicine Information. Otherwise, the letter contained nothing of apparent relevance to the present proceeding.
272 The final “Dear Pharmacist” letter was dated 6 September 2004 and is, for present purposes, somewhat more controversial than the other four. It was over the hand of the Medical Director of MSDA, and read as follows:
You may have seen or heard media coverage this week of a story about the risk of heart attack occurring in patients taking VIOXX® (rofecoxib).
More than 84 million patients worldwide have been prescribed VIOXX since the product first became available in 1999. VIOXX has been extensively studied with more than 24,000 patients being treated with VIOXX in randomized, controlled clinical trials. Based on all the data available, Merck stands behind the efficacy, overall safety profile and cardiovascular safety profile of VIOXX.
A number of health agencies around the world have reviewed the data on Cox-2 inhibitors and have drawn similar conclusions – these agencies include the Therapeutic Goods Administration (TGA) and the Pharmaceutical Benefits Advisory Committee (PBAC) in Australia, the Food and Drug Administration in the USA, and the Committee for Proprietary Medicinal Products at the European Commission.
In April 2004, the European Commission announced its adoption of an opinion confirming that Cox‑2 selective inhibitors, including VIOXX, have a positive balance of benefits and risks based on a comprehensive review conducted by the Committee for Medicinal Products (now referred to as the Committee for Medicinal Products for Human Use). This opinion was supported by an extensive analysis of data provided by Merck confirming the overall safety profile, including gastrointestinal and cardiovascular tolerability of VIOXX. The analysis was based on a review of 27 VIOXX studies.
Also this year, the Australian PBAC completed its two‑year review of Cox-2 selective inhibitors. They concluded that the data do not support an increased risk of cardiovascular tolerability for patients on rofecoxib (VIOXX) compared with traditional NSAIDs.
The Product Information for VIOXX states: “The difference in antiplatelet activity between some COX-1 inhibiting NSAIDs and COX-2 selective inhibitors may be of clinical significance in patients at risk of thrombo‑embolic events. Physicians should assess the importance of these data for an individual patient at risk for cardiovascular thrombo‑embolic events, if considering long term therapy with a Cox‑2 selective inhibitor.”
Patients at risk of cardiovascular disease should be on low dose aspirin. This is independent of whether they are taking a Cox-2 selective inhibitor such as VIOXX or Celebrex or a traditional NSAID.
The Product Information for VIOXX states that “VIOXX can be used with low dose aspirin. At steady state, VIOXX 50 mg once daily had no effect on the anti‑platelet activity of low-dose (81 mg once daily) aspirin, as assessed by ex vivo platelet aggregation and serum TXB2 generation in clotting blood. In 3 six‑week, double‑blind, randomised clinical trials, VIOXX 12.5mg or 25mg was administered to osteoarthritis patients concomitantly with low-dose aspirin (less than or equal to 325mg daily) (N = 161). No clinically important differences were noted for users of VIOXX plus aspirin versus VIOXX alone in the overall incidence of clinical adverse experiences. However, concomitant administration of low dose aspirin with VIOXX may result in an increased rate of GI ulceration or other complications, compared to use of VIOXX alone. Because of its lack of platelet effects, VIOXX is not a substitute for aspirin for cardiovascular prophylaxis.”
Finally, it should be noted that the major finding from the VIGOR study, a prospective study designed to evaluate the gastrointestinal safety of this new class of therapy in comparison to the traditional NSAID, naproxen, was that VIOXX was associated with significantly fewer clinically important upper and lower gastrointestinal events. This was the prespecified endpoint of the VIGOR study.
The PBAC review into Cox‑2 inhibitors determined that VIOXX does have substantial advantages in gastrointestinal tolerability compared to traditional NSAIDs.
Again, a copy of the Product Information was included with the letter. As pointed out by the respondents, this letter was not the subject of evidence by any witness. It was not put to any of the respondents’ witnesses. It was listed, without further comment, in the applicant’s final submissions as an instance of MSDA having failed to provide any adequate information, advice or warning to health care professionals that consumption of Vioxx materially increased the risk of suffering the pleaded cardiovascular conditions until the withdrawal of the product in October 2004.
273 The position ultimately reached in relation to the “Dear Doctor” and the “Dear Pharmacist” letters is that I would find that, with the exception of that dated December 2001, those letters were sent to doctors and pharmacists in Australia generally as apparently intended. Whether any particular letter was received by the doctor or pharmacist whose position may become relevant to the circumstances of a particular group member is, however, another question, the answer to which cannot be regarded as conclusively tied to that finding. My provisional view is that the finding would in effect place the evidentiary onus on any party who desires to contend that, in relation to a particular doctor or pharmacist, the position is not as it appears to be. I make an exception in the case of the letter dated December 2001 because the finding I make is that it was not sent: any further debate on the question whether it was received by a particular doctor would be inconsistent with that finding.
274 Another important means by which MSDA communicated with general practitioners involved the use of senior members of the profession. Ms Dobson said that the use of speakers as part of an “education cascade” or a “communication cascade” was a commonly-used approach by MSDA and other companies. By that she meant, for example, that MSDA would have a small group of usually very senior physicians who had been involved in clinical trials with a particular product. There would then be a slightly larger group of senior physicians who were, perhaps, specialists in the area. The next level down would be other specialists, after which there would be general practitioners. Asked how the senior physicians were identified, Ms Dobson said that that was usually done by MRL. Then the other senior physicians would almost “self‑select in a therapeutic area”, in the sense that they were usually very eminent, were very well‑published, and held important university appointments. Asked how the information and opinions would “cascade to the rest of the profession”, Ms Dobson said that MSDA would often ask the senior physicians if they were prepared to be part of an education program, such as by acting as speakers, by authoring slide kits, or by speaking to groups (large or small) of general practitioners. She agreed that the ultimate objective was “to get word about the product out to the broadest market, within the profession”.
275 In 1999, MSDA established an Arthritis Advisory Board. That companies such as MSDA should have such boards seems not at all uncommon. Dr Bertouch (to whom I refer in para 277 below), for example, either had been or was (ie at the time of giving evidence) a member of nine industry advisory bodies, including that established by MSDA. The Board was to consist of 11 key specialists from around Australia, and had the following objectives:
– To ensure ongoing high profile for VIOXX in specialist arena
– To ensure a group of highly trained specialists are available to communicate VIOXX messages other specialists and GP’s.
– To help to block Searle/Pfizer activities
– To assist with advising MSDA on other issues as required.
The applicant submitted that the Board was “part of the marketing budget” and, whether or not that was so as a matter of MSDA’s internal accounts, on any view significant effort was put into the education of Board members apparently with a view to them being well‑informed about Vioxx for the achievement of the above objectives. According to the Vioxx “launch” marketing plan for 1999‑2000, the first official meeting of the Board was to be held on 31 July 1999, and after which −
… the entire Advisory Board will be sponsored to travel to the William Harvey Research Conference on the “Potential of COX-2 Inhibition” to be held in Lisbon, Portugal from the 18th to the 20th October 1999. This will then be followed by a visit to MRL to meet with some of the key VIOXX researchers. This activity is designed to fully prepare the Advisory Board for the launch of VIOXX and to gain their ongoing support for MSDA into the future.
276 The applicant submitted that, in addition to the objectives referred to above, there were “unofficial objectives” which MSDA had for the Board. In this regard, he relied on an email said to have been sent by Ms Annette Wakeford on 10 June 1998. Ms Wakeford was a product manager with MSDA, and she did send an email on that day. However, hers was an insignificant email in response to a more important one from Mr Sumeet Sud, the Asia Pacific product manager for Vioxx. It was Mr Sud’s email that proposed the adoption of “unofficial objectives”, but they were not for the Advisory Board in Australia. They were for an Asia Pacific Advisory Board which had just then been established. There was no evidence that MSDA harboured such unofficial objectives for the Australian Board. Indeed, when Mr Sud’s list of objectives was put to Ms Dobson, she said that she was unaware of it, and she strongly disapproved of the notion of having unofficial, uncommunicated, objectives in the case of the Board. She said:
I don’t think Merck … – the way we conduct ourselves or the way we try to conduct ourselves [–] would support this. Sumeet put this down. I don’t think many people would have supported having something unofficial. … What I am reacting to is the sort of notion that this is unofficial. This could have been discussed quite openly with them and they would have either agreed or not, so whether they did these things or not is their choice in some cases.
In the circumstances, I am not prepared to find that MSDA had the unofficial objectives for the Australian Board as proposed by the applicant
277 Two members of the Board were called by the respondents. The first was Dr Jim Bertouch, consultant rheumatologist and Chairman of the Department of Rheumatology at Prince of Wales Hospital, Sydney. He answered questions about the public statements he had made on coxibs. He agreed that he came to view Vioxx as a highly specific COX-2 inhibitor that offered efficacy comparable to traditional NSAIDs and greater gastrointestinal safety. He stated that proposition publicly at lectures, forums and the like, not in the sense of talking about Vioxx as such, but in the sense of talking about the whole range of anti‑inflammatory medications that were available, and what the advantages or disadvantages of each might have been. However, he considered that Vioxx was better than Celebrex with respect to gastrointestinal tract outcomes, and said so at symposia, conferences and the like. He wrote positive articles about coxibs as a class of drug, and accepted that it was likely that he would have said that Vioxx was superior to Celebrex in those ways. He never sought to persuade practitioners that Vioxx ought to be part of their formularies. He was never asked by MSDA to make public statements, media statements or the like in relation to Vioxx. He was also on the Celebrex advisory board, and did not remember “singling out one agent and promoting that agent or in some other way putting it across as being the agent that I thought was the best one …” He did not recall being involved in training sessions for MSDA representatives, but allowed that he may have done so. Asked whether he was invited to speak to practitioners at dinners, for example, and to make presentations concerning Vioxx, he replied:
I do a lot of talks for GPs. In fact, that’s part of the process, I suppose, of being appointed to a teaching hospital, that one would talk to groups of GPs in the local community, apprise them of what was happening in the field, but one would not do a presentation just about a single product. One would always balance that with whatever else was in that field. So if I did talks to GPs about anti‑inflammatory agents or to medical students or to people training to be physicians, it would have been usually across the board talking about the broad area of those things rather than an individual agent.
Pressed to recall whether, because of his views about the gastrointestinal superiority of Vioxx, “those present would have understood that you thought Vioxx was the best choice in that area”, Dr Bertouch said:
Yes, but I guess there would be some qualifications for that because Celebrex was also a good agent to use as far as anti‑inflammatory effect was concerned and it did show almost a gastrointestinal advantage over its comparators but didn’t quite get there. So it wasn’t that it was no good but it wasn’t quite as striking in the sense of the results, so far as the gastrointestinal tract was concerned.
278 The other member of the Board called by the respondents was Prof Neville Yeomans, Foundation Dean of Medicine, University of Western Sydney. He agreed that he had given speeches at meetings, symposia and the like concerning the increased gastrointestinal safety of Vioxx. He did not provide information or advice to regulatory authorities or pricing authorities on behalf of MSDA, but there were instances in which he, as a member of the Board, participated in discussions on such questions as, for example, whether certain data were “robust in terms of [gastrointestinal] safety”. He wrote “at least two or three articles … about Vioxx”. He was not aware whether MSDA had used the things he had written for promotional purposes, but he accepted that it may have. He talked at a number of meetings – often in the context of NSAIDs generally rather than of robecoxib as such – about ways of protecting against the damage to the stomach. He did so “in many fora and overseas meetings”. He provided a gastrointestinal opinion for inclusion in a CD‑ROM which featured Prof Weir, who was brought to Australia by MSDA. He did not recall having provided training to MSDA representatives. In re‑examination, Prof Yeomans denied that he was acting as the mere mouthpiece for MSDA. Indeed, the way he responded to the question which was designed to elicit that denial gave me the distinct impression that he felt a sense of affront that anyone might consider that he was so acting. He said that, as an academic, his job was to teach people and that, like any specialist, he had a job to teach his colleagues in his area of specialty. It was not suggested on behalf of the applicant that I should reject this evidence.
279 The opinions of members of the Board were also used by MSDA in “advertorials” on occasions. There were three examples of such advertorials in evidence (in referring to them as “examples”, I do not mean to imply a finding that there were others − the evidence would not permit the making of such a finding). They all appear to have been produced in the period around October/November 2001. Save in one respect, the applicant’s submissions went no further than to note that these advertorials confirmed “statements supportive of Vioxx” made by the Board members concerned. The exception related to a statement by A/Prof Henry Krum, Head of the Clinical Pharmacology Unit at the Department of Medicine, Alfred Hospital (but said in the applicant’s submissions to be a cardiologist) that Konstam (2001) was “a more rigorous meta‑analysis of rofecoxib trials” than Mukherjee (2001). I did not understand the applicant, however, to have proposed that A/Prof Krum did not in fact hold that view, or that it was a view which was not legitimately open to him or to anyone else. Reference to the advertorials did not advance the applicant’s case against MSDA.
280 Although the applicant made much of what he proposed was the use of senior specialists and other opinion leaders in the profession as part of MSDA’s promotional efforts for Vioxx, his submissions focused upon process rather than content, as though I should consider that the existence of such a modus operandi on the part of MSDA was of itself indicative of culpability apropos the causes of action on which he sued. There was, however, no evidence of any actual communication made by any of these specialists or opinion leaders that was other than a conscientious statement of his or her views as a scientist made independently of the commercial interests of MSDA. I accept that the enrolment of such specialists and opinion leaders was done by MSDA to spread the good word, as it were, about Vioxx among the prescribing fraternity, but this practice was quite innocuous apropos the applicant’s causes of action, and seems to have been regarded by the people concerned as legitimate and conventional for a drug manufacturer such as MSDA.
281 The next important means by which MSDA communicated with general practitioners, and the means which most obviously involved sales and marketing as such, was by way of its sales representatives. The MSDA representatives visited doctors’ rooms to advance the cause of Vioxx (and of other products supplied by MSDA) in face‑to‑face discussion. Since doctors themselves did not (at least in the typical situation) purchase Vioxx, such representatives were not engaged in direct selling, but their broad purpose was to influence the prescribing practices of those whom they visited, and their role was, in that sense, effectively that of sales representatives.
282 There seems generally to have been every attempt made to ensure that a consistent marketing message was conveyed about Vioxx (and, I infer, about other drugs being marketed by MSDA). Mr McAuley said that sales representatives had various different categories of resource materials provided to them by MSDA. A standard form of document provided to the representatives was the “Detail Aid”, described by Mr McAuley as a brochure – which might include graphs or charts based upon clinical studies − designed to be used by representatives as a framework for discussion with health care providers, usually containing visual aids to help explain product information. Mr McAuley said that detail aids were not intended to be given to or left with practitioners, but Ms Dobson said that they might be, depending on the choice of the representative concerned. On any view, detail aids were intended to be shown to practitioners, at least to the extent necessary to facilitate the understanding of the points being made by representatives from time to time. If the respondents’ schedule of evidence is to be believed, there were 39 detail aids marked as exhibits in this proceeding.
283 By reference to MSDA marketing plans, the applicant submitted that detail aids were much more than as described by Mr McAuley and Ms Dobson. For example, the plan for 2003 (prepared in November 2002) had a section headed “Tactics To Influence Brand Choice – Choose Coxibs, Initiate VIOXX And Switch To VlOXX”. Within that, there was a sub‑section headed “Detail Aid For Switching To VIOXX, Targeting Medicated But Not Dissatisfied Patients (Patient I) – Musculoskeletal team”. The purpose of the detail aid was described as follows:
The detail aid will present data that aims to provoke the doctor to re‑examine his [osteoarthritis] patients and assess their level of satisfaction with their current medication. Patient photographs will be used to facilitate the doctor thinking about actual patients rather than focussing on clinical data. Breakthrough pain and/or GI safety and tolerability will be utilised as the driving reason behind the need to reassess patient satisfaction. Again, satisfaction will be the central message for VIOXX with strength, safety and simplicity being drivers of the satisfaction.
However, the applicant did not draw attention to any statement in a detail aid as such which was false or misleading. Although the above text from the marketing plan, and similar statements in some other material MSDA documents, suggest that practitioners should have the “safety” of Vioxx impressed upon them, in none of the examples admitted into evidence did a detail aid go further that to proclaim the gastrointestinal safety of Vioxx. Otherwise, the applicant’s complaint about the detail aids was that they omitted to refer to cardiovascular risk, and made no mention of VIGOR. This is true, but the circumstance is relevant to the applicant’s allegation that MSDA failed to warn, an allegation that was separate from his criticism of MSDA’s marketing campaign.
284 The next category of marketing material referred to by Mr McAuley was the “leave‑behind” card. He described it as typically a double‑sided card that included information about a specific aspect of an MSDA medicine. It was designed to convey information in a concise format, and to serve as a ready reference for the practitioner. As the name implied, such a card was intended to be left behind with the practitioner. No submission of the applicant was based upon any leave‑behind card.
285 Another document that was produced by the marketers at MSDA was the “Objection Handling Module”. Mr McAuley explained that the purpose of this document was to enable sales representatives nationally to respond to concerns raised by health care professionals with consistent and accurate information. Under cross‑examination, Mr McAuley explained the intent of objection handling modules as follows:
The intent of this is to ensure that a representative truly understands the doctor’s questions so they can answer those in the most appropriate way with the most appropriate materials, discussion points, whatever they need. Often you will hear a question that once you seek to understand that question a little further you get to understand the best way to answer the question for that doctor so that you actually can address potentially an underlying concern as opposed to a superficial question and that way you can either get to the end point of agreeing or disagreeing with a doctor; you can agree to disagree over the outcome of a discussion but it allows you to make sure that you are addressing that specific request. … [I]f we are able to ensure we make best use of their time with those doctors without wasting it then the objection handling model is a fantastic way to make sure you understand the question being asked properly before you provide an answer.
286 What Ms Dobson described as an “early” objection handling module was organised by reference to a series of questions, or objections, that might be posed by a general practitioner, for example. They were:
1. “I’ve already got Celebrex. It works and I’m comfortable with it. Why should I use Vioxx?”
2. “What about the problems that Vioxx has with an increase in cardiovascular events as shown in your own large outcomes trial (VIGOR). There were no such problems with Celebrex in their study (CLASS)”.
3. “Vioxx has more renal problems than Celebrex.” Or
“Vioxx has more problems with causing hypertension than Celebrex.” Or
“Vioxx has major problems with oedema and much more than Celebrex”.
4. “There are some problems with Vioxx. After all, it doesn’t have an indication in rheumatoid arthritis because of its drug interaction with methotrexate.”
5. “VIOXX – another sulfur drug! Why create another COX-2 that is contraindicated for patients with ‘sulfa’ allergies?”
6. “What about the serious drug interaction between VIOXX on warfarin?”
7. “How does VIOXX compare to NSAIDs?”
It is the second objection set out above which is of present concern. The advice to the representative was, first, to clarify where the doctor had heard this information, and to clarify the “real concern” from the doctor. The following was then set out:
Doctor, this is just mischief and misinformation created by Representatives of the other COX-2. Vioxx has no problem with cardiovascular disease compared to conventional NSAIDs or the other COX-2. In all clinical trials with Vioxx, overall mortality and cardiovascular mortality is low and comparable to conventional NSAIDs. Of course, Vioxx is not a substitute for low‑dose aspirin, as it has no effect on COX-l in humans even at up to 80 times the dose.
Should the doctor require further clarification, the representative was advised to point out that the lower number of myocardial infarctions observed in the VIGOR trial in the case of patients taking naproxen was consistent with the COX-1 selectivity of that drug, and with its aspirin‑like ability to block platelet aggregation. It was said that Vioxx, like all COX-2 inhibitors, did not block platelet aggregation, and therefore would not be expected to have similar effects. If the doctor required even further clarification, the representative was advised to inform the doctor that the incidence of myocardial infarctions in the VIGOR trial was 0.4%, that the like figure over “the entire VIOXX clinical studies database of over 5,000 patients” was 0.6% (said to be similar to placebo and comparator NSAIDs), and that the like figure with celecoxib in the CLASS trial was 0.5%. The document continued:
Based on these facts and figures, it is fair to say that VIOXX does not cause cardiovascular disease and that there is no difference between VIOXX, the other COX-2 [ie celebrex] or conventional NSAIDs.
287 The objection handling module just referred to was the subject of a letter of complaint to MSDA from Pharmacia Australia Pty Ltd (the manufacturer of Celebrex) dated 16 February 2001. The comparisons which placed Vioxx in a favourable light appropos Celebrex were objected to, as were the statements that the rate of myocardial infarction in VIGOR was similar to that reported in other studies, and that the VIGOR results were explicable by the reference to the cardioprotective effect of naproxen. As to the latter, it was pointed out in the letter that such an effect had never been shown in the public literature, and that the manufacturers of naproxen had publicly stated that they were unaware that their product had that kind of an effect. It seems that, from about this time, there was much mutual disparagement by MSDA and Pharmacia of each other’s coxibs in the marketplace. The evidence contains a number of examples of exasperations being expressed by sales and marketing people at MSDA about the practice of Pharmacia representatives pushing the line, as it were, that the consumption of Vioxx was associated with cardiovascular risk. For their part, the MSDA representatives likewise had criticisms of Celebrex. Ultimately, a meeting between senior management from the two companies in September 2001 reached a loose consensus that they would not disparage each other’s products. As minuted by Mr Back, there was general agreement that –
both products have obvious superiority over NSAIDs. We should be looking at other issues, rather than putting down both products. Market is difficult at this time, pressure from PBAC over pricing, fighting each other may not be in either companies [sic] interest at this time. We also need to have a close look at how we promote the class with MOBIC about to enter.
The companies also agreed “to only include comparisons to NSAIDs on the [gastrointestinal] issue”. There are suggestions in the evidence that these agreements were fragile, and that MSDA at least was noticing a resumption, to some extent, of the disparagement of Vioxx on cardiovascular-related grounds by about the middle of the following year.
288 The applicant also drew attention to an objection handling module produced MSDA in response to Mukherjee (2001) which had been published in August 2001. If a practitioner were inquiring about the problems that Vioxx had with an increase in cardiovascular events, as shown in the Mukherjee article, the sales representative was advised to provide the following “answer”:
Dr I can understand your concern however let me reassure you that when you consider all of the available data, there is no difference in cardiovascular events between Vioxx and placebo. Vioxx is an appropriate and efficacious therapy for your patients with osteoarthritis. Dr, with regard to this article it is important to note that:
1. Compelling evidence is available that confirms that there was no difference in the overall incidence of cardiovascular events among patients taking Vioxx and placebo and between patients taking Vioxx and other non‑naproxen NSAIDs
2. Vioxx can be used in patients at risk of cardiovascular disease. Importantly Vioxx is not a substitute for aspirin for cardioprotection
3. Even the authors have conceded their analysis has “several significant limitations”.
The representative was advised to inform the doctor:
Compelling data from a rigorous meta‑analysis of data from 19 controlled clinical studies with over 28,000 patients showed there was no difference in the overall incidence of cardiovascular events among patients taking Vioxx and placebo and between patients taking Vioxx and other non‑naproxen NSAIDs.
The doctor was to be informed that the results of VIGOR were consistent with the aspirin‑like properties of naproxen, and to be reminded that aspirin itself was not allowed in the study. The doctor was to be informed that COX-2 inhibitors may be used in patients at risk for cardiovascular events, but reminded that the prescribing information for both Vioxx and Celebrex stated that they should be used with caution, and should be introduced at the lowest possible doses in patients with concomitant conditions such as hypertension or heart failure. That information (again, for both products) also clearly stated that they were not a substitute for aspirin for cardio‑protection. The document concluded as follows:
Extensive cardiovascular data already exist on Vioxx and these data – which were not incorporated into the authors’ analysis – suggest that there is no increase in the risk of cardiovascular events as a result of treatment with Vioxx.
289 I shall refer also at this stage to the MSDA marketing plans that were developed during the period that is relevant to the present case. I do so merely to give an impression of the headline marketing messages which were proposed by those plans, and to touch upon the way that issues of safety, and of cardiovascular safety in particular, were dealt with. As I explain in para 858 below, however, evidence of the existence of such plans, and the formulation of those messages, can be regarded as little more than a necessary foundation for later evidence about the communications in fact made consistently with them. For that reason, what I set out below is intended to be indicative rather than exhaustive, and to leave open the prospect that any aspect of the evidence relating to the applicant’s marketing case may be the subject of specific consideration in the context of a particular group member at the appropriate stage.
290 As Marketing Director after June 2001, Ms Dobson was responsible for Vioxx strategy, a key aspect of which involved the approval of Vioxx marketing plans. She identified the marketing plan for the launch of Vioxx in Australia in February 2001. In that document, the “Key Promotional Messages” were:
– Strength
Vioxx has powerful 24‑hour relief of OA symptoms without breakthrough pain.
Vioxx has the power of maximum recommended doses of NSAIDs.
– Simplicity
Vioxx offers benefits over celecoxib:
o 24 hour pain control in a once daily dose for all patients
o no sulfonamide contraindication
o the cytachrome P450 is not the dominant metabolic pathway
o no dosage adjustments are necessary
– GI safety
Serious GI complications (PUB) with Vioxx were significantly less than the conventional NSAIDs ibuprofen and diclofenac.
291 These promotional messages were not, however, the only things dealt with in the marketing plan. In the executive summary, a number of issues that would need to be resolved in connection with the competition from Celebrex were mentioned. One of them was to minimise the impact of the portrayal by the distributors of Celebrex “of the renal and CV profile of Vioxx compared to Celebrex”. The plan contained a “SWOT” analysis, the first entry under the “weaknesses” section of which was: “The perceived safety issues with renal and CV profile”. In the section of the plan dealing with “issues”, and in the subsection thereof dealing with “marketing”, the following appeared:
Renal and cardiovascular (CV) profile: The renal and CV profile of Vioxx is similar to Celebrex and other NSAIDs. However, Pharmacia/Pfizer are using unpublished data and/or studies that are poorly designed to create the impression that there are renal and CV issues with Vioxx. This has proved a very successful ploy is the USA, where market research has shown that while the majority of physicians do not differentiate between Vioxx and Celebrex on CV and renal safety, but when they do they favour Celebrex.
The marketing plan contained a “tactical plan” which had been developed to address the key issues that Vioxx would face in 2001. One of the issues was “renal/CV profile and efficacy profile”. A budget was laid out for a number of items, including sales aids, detail aids, brand name reminders, direct marketing, extra call programs, GP evening meetings, GP weekends, teaching faculty, registrar weekend, ARA symposium, Advisory Board and CV advisory group.
292 In April 2002, Ms Dobson approved the Vioxx marketing plan for that year. In the section headed “Marketing Strategy”, there was a subsection headed “Positioning Statement”. Here it was said that, for doctors treating patients suffering from chronic pain and inflammation, Vioxx was the solution “to unsurpassed ‘power without compromise’ that secures a brighter future for you and your patients”. It was said that Vioxx provided doctors with the power and control to give their patients independence and freedom, with the peace of mind that comes from giving their patients “the safety they deserve”, with the ability to secure their patients’ confidence by appropriately managing their expectations, and with recognition as healers. These benefits were said to arise by reason of a number of features of Vioxx, including “Vioxx GI Safety Profile and GI Tolerability are both superior to traditional NSAIDS (VIGOR outcome study)”, and “meta‑analysis confirms CV safety of Vioxx (Konstam)”. It was also pointed out that “Vioxx safety has been proven over the long term through 19 studies involving 28,000 patients”. Although not mentioned, this seems to have been a reference to Konstam (2001). (As it happens, Konstam (2001) was concerned with 23 studies involving more than 28,000 patients, but drafts of the article discovered by Merck referred to 19 studies. I note also that one of the applicant’s expert witnesses, Prof Jelinek, agreed with cross-examining counsel that there were 19 referred to in Konstam (2001).) In the section of the marketing plan headed “Marketing Program”, there was a table headed “Summary of promotional tactics by leverage points and behavioural objectives”. One of many entries alongside the leverage point “brand choice – prescribe Vioxx” was “CV mailer”. The text associated with that entry noted that it was an objective to inform doctors that a large scale meta‑analysis showed that Vioxx was not associated with an increased cardiovascular event risk; rather, it was naproxen which reduced the risk. It was said that mailers carrying the meta‑analysis data and safety messages on cardiovascular effects would be mailed to general practitioners. Dealing with the subject of “market definition”, the marketing plan noted that there had been “a series of negative reports on renal and cardiovascular safety concerns regarding coxibs sent May 2001”.
293 In November 2002, Ms Dobson approved the Vioxx marketing plan for 2003. In the section dealing with “Market Definition”, the “negative press regarding around [sic] safety issues (cardiovascular and renal)” was noted. Likewise, what were described as “residual safety concerns (cardiovascular and renal)” were identified as one of the “key challenges” for Vioxx in 2003. The benefits of Vioxx as they should be seen by doctors, and the reference to Konstan(2001) as confirming the cardiovascular safety of Vioxx, were retained from the 2002 marketing plan.
294 There was a further group of submissions made on behalf of the applicant about the marketing of Vioxx. They were grouped under the heading “Disseminating the Marketing Messages − Journals and other Media”. It was submitted that the respondents − and here the applicant made no obvious discrimination as between Merck and MSDA − caused articles to be written in medical journals by which the benefits of Vioxx would come to the attention of the prescribing community. These submissions, however – and the evidence on which they were based − proceeded at a very high level. They were far removed from the circumstances of any group member, including the applicant. There was no evidence that anything written in any journal influenced the prescribing decisions of Dr Dickman, for example. Neither, when he came to that part of his case which related to the acts and omissions of MSDA which were claimed to be negligent and to amount to misleading conduct, did the applicant draw my attention to anything of present relevance written in a medical journal. The respondents submitted that there was in effect a disconnect between the applicant’s marketing allegations and the chain of events that led him to consume Vioxx. At least in relation to journals, I consider that there is substance in this criticism.
295 The section of the applicant’s written submissions to which I have just referred was based substantially on the evidence of Prof Donovan and of Prof George Jelinek (a medical practitioner with expertise in the area of medical publications − what he described as “journalology”). The factual basis of that evidence lay in documents provided to them by the applicant’s solicitors (such documents having been, I presume, discovered by the respondents). These witnesses did not, however, propose that anything written in a journal by a contributor associated with the respondents was wrong, or should not have been written. To the extent that the evidence dealt with particular articles about Vioxx, and to the extent that criticisms of the content of those articles were part of the applicant’s case, I deal with them elsewhere in these reasons. The evidence of these witnesses was concerned more with the respondents’ practices in the way they used journal articles as a means of promoting Vioxx, and with what was said to be a departure from customary professional norms in that respect. I am not, however, concerned to try the respondents for transgressions of this kind. In some instances also these witnesses examined emails (provided to them by the applicant’s solicitors) which preceded the submission of articles for publication, with a view to comparing what Merck knew at the time with what its authors chose to publish. To the extent that the applicant relied on particular instances of this, I deal with them elsewhere. Generally, I am not persuaded that the evidence of Prof Donovan or of Prof Jelinek about journals, and the publication practices of the respondents, makes any material contribution to the applicant’s case.
296 I should also mention a publication of MSDA called “The Australian Journal of Bone and Joint Medicine”. The applicant described this as “a fake medical journal”. It had the setup of a peer-reviewed, independent, journal, and much of the material in it had been written by professional contributors (whether or not acknowledged). However, it was neither peer-reviewed nor independent. It was effectively a highbrow means of promoting some of MSDA’s products (including Vioxx) within the medical profession. The journal ostensibly had an editorial board, but there is reason to be sceptical about the reality of that institution: Dr Bertouch was named as a member of it, but he was unaware of that membership, and had never attended a meeting of the board. MSDA led no evidence of its own about this journal. However, notwithstanding these reservations, the applicant did not draw my attention to anything published in this journal that was wrong or misleading, or that tended to substantiate any of the allegations that made up his causes of action against MSDA. Neither was there any evidence that this journal had come to the attention of any practitioner whose opinions contributed, directly or indirectly, to the applicant’s consumption of Vioxx.
part iv − vioxx and the risk of cardiovascular disease
Introduction
297 Central to the applicant’s case were his allegations that the consumption of rofecoxib, and therefore of Vioxx, materially increased the risk of suffering the pleaded cardiovascular conditions, and that the respondents knew or ought to have known of that circumstance. Consistently, substantial sections of the applicant’s submissions were devoted to establishing, first, that there was such an increase in risk, and, secondly, that the respondents knew or ought to have known of it. In the first section of his submissions, the applicant relied upon the current state of scientific understanding, drawing attention to a number of sources of information and opinion running well beyond the date when Vioxx was withdrawn from the market. In the second section, the focus was on the unfolding science as it existed from time to time until the withdrawal, but with an emphasis on the proposition that the respondents at least ought to have known of the risk by about the end of the first quarter of 2000.
298 The concept of “risk” is, of course, much used in personal injury litigation, especially in product liability cases. The relatively uncomplicated denotation of that word, however, conceals a veritable thicket of connotations, which I have little option but to penetrate with a view to explaining the kind of “risk” to which the applicant refers in his submissions in this case. In some situations, for example, a “risk” refers to the danger that something of conspicuously obvious causation will follow upon some injudicious course of conduct, such as the risk of injury from jumping from a moving train or diving into shallow water. In other situations, a “risk” refers to the likelihood of events or influences acting or taking place in a way which would be regarded as unwanted, such as the risk of it raining when one ventures out on a cloudy day without an umbrella, or the risk which a motorist takes when he or she fails to return to his or her parked vehicle within the allowed time.
299 Closer to the facts of the present case, it seems that there may be some situations in which surgery involves inherent risks. Apart from the prospect of something going wrong, or the patient having an allergic reaction to a drug or anaesthetic, there is the classic case, of which Rogers v Whitaker (1992) 175 CLR 479 was an example, it seems, where every so often some unwanted outcome results from a particular procedure. Here I have in mind a situation in which it is established, as a matter of medical science, that such an outcome will occur, albeit very infrequently. The “risk” which the patient takes, when properly informed, is the risk that he or she might be one of the unlucky few. As a matter of common experience, one would think that, if the surgery were going to give the patient a significant improvement in his or her wellbeing in some respects, he or she might well decide to take a very small risk of the unwanted outcome occurring (as the trial Judge held would have been the case, had a warning been given, in the matter which reached the High Court as Rosenberg v Percival (2001) 205 CLR 434).
300 In the present case, in what I have described as the first section of the applicant’s submissions on risk, he alleges that sufficient is now known about the consequences of consuming Vioxx to justify the conclusion that it leads to an increase in the risk of suffering the pleaded cardiovascular conditions. He limits his case to an allegation of an increase in risk, because he accepts that such conditions occur from time to time in any event, so there will always be a risk of them. He uses the word “risk” in the sense of alleging that there was − either as a known fact or as something that may reliably be inferred – a feature of the operation of Vioxx in the vasculature that caused or contributed to the occurrence of the pleaded cardiovascular conditions. That is to say, the applicant alleges that we now know that Vioxx had such a tendency, and that anyone who took it was exposed to the “risk” that the tendency would come home, as it were.
301 In this part of my reasons, I am concerned with whether the consumption of Vioxx involved an increase in risk in this sense. I am not here concerned with the question whether, in an environment in which the existence of that actual risk was unknown to science, or was attended by uncertainty, the respondents, by continuing to market Vioxx, were exposing their customers to the “risk” that it might, ultimately, be found to be harmful. That question is the subject of a later part of these reasons.
302 The applicant’s case was opened substantially by reference to the FitzGerald hypothesis, but, in closing, counsel relied primarily on the results of the APPROVe trial for their submission that the consumption of Vioxx in fact contributed to the occurrence of the pleaded cardiovascular conditions. They submitted that, since APPROVe, the existence of the risk was no longer the subject of any realistic scientific uncertainty, or at least should be found to be established on the balance of probabilities.
303 The applicant submitted that this conclusion based on APPROVe derived support from what was described as “other clinical trial evidence”. In this respect the applicant relied on the results of the VIGOR trial, upon the arthritis and Alzheimer’s studies conducted by Merck, upon the data available from the terminated VICTOR study, upon the meta‑analyses of Kearney (2006) and Juni (2004) and upon other coxib studies to which I shall refer. It was submitted that the conclusion derived support from such evidence as there was of a plausible physiological mechanism by which rofecoxib contributed to vascular disease, namely, the FitzGerald hypothesis, the progression of atherosclerosis and the development of hypertension. It was submitted also that the decision in September 2004 to withdraw Vioxx from the market demonstrated an appreciation by Merck itself of the risk alleged by the applicant and that a vote of the FDA Advisory Committee in February 2005 (to which I shall come in due course) provided additional support for that allegation. The applicant submitted that the elevated cardiovascular risk was immediate (in the sense of arising promptly upon a patient commencing a course of Vioxx) and enduring (in the sense of existing well after a patient had discontinued taking Vioxx). It was submitted that the risk was particularly evident in the case of patients who might normally be expected to be taking drugs for the relief of arthritic pain.
Overview of expert opinion
304 Probably the single most significant circumstance contributing to the length and complexity of the present litigation is that there is no scientific consensus with respect to the matters referred to above. That lack of consensus is reflected in the conclusions of the scientists called by the parties at trial. The applicant called −
· A rheumatologist, Prof Leslie Cleland, Director of Rheumatology at the Royal Adelaide Hospital;
· Two biostatisticians, Prof David Madigan, Chair of Statistics at Columbia University in New York City and Prof Mark Woodward, Director of the Epidemiology and Biostatistics Division of the George Institute for International Health (since 2006, on leave of absence, working at the Mount Sinai School of Medicine in New York); and
· Two cardiologists, Prof Richard Harper, Emeritus Director of Cardiology at Monash Medical Centre, a Professor in the Department of Medicine at Monash University and Clinical Lead in Cardiology in the Department of Human Services, Victoria, and Prof Douglas Zipes, Emeritus Professor of Medicine at Indiana University and former Director of the Division of Cardiology at the Krannert Institute.
The respondent called −
· A rheumatologist, Dr Jim Bertouch, Chairman of the Department of Rheumatology at Prince of Wales Hospital, Sydney;
· A biostatistician, Dr Ronald Marks, until 2004 a Professor in the Department of Statistics at the University of Florida; and
· Two cardiologists, Prof David Celermajer, Scandrett Professor of Cardiology at the University of Sydney and Clinical Director of the Heart Research Institute, and Prof Douglas Vaughan, Irving S Cutter Professor and Chairman of the Department of Medicine at the Northwestern University Feinberg School of Medicine, Chicago, and Physician-in-Chief of Northwestern Memorial Hospital.
305 Notwithstanding that these experts conferred (grouped according to their respective specialisations), there remained fundamental disagreement between them. I shall commence by noting the extent and nature of that disagreement.
306 In his report, Prof Zipes surveyed the published research, and the results of clinical trials, and expressed the following conclusion:
It is my opinion that the experimental and clinical evidence strongly support the conclusion that COX-2 inhibition, and specifically Vioxx, increases the risk for cardiovascular complications, particularly when there may already exist some degree of coronary disease or risk factors.
The other cardiologist called by the applicant, Prof Harper, said:
There is now overwhelming scientific evidence from randomised studies, from meta‑analysis of randomised studies and from observational data that in prone individuals, regular ingestion of Vioxx substantially increases the risk of adverse vascular events such as heart attack. … To my knowledge there is no credible scientific evidence to suggest otherwise. … There is some variation in the scientific literature as to exactly what the degree of increased risk is and how long one needs to take Vioxx before the risk becomes apparent. My interpretation of the scientific data is that the risk associated with Vioxx starts within the first month of initiation of therapy and occurs with both 25mg and 50mg doses, but the risk is higher with the 50mg dose.
307 The biostatisticians called on behalf of the applicant expressed similar conclusions. Prof Woodward examined, and where necessary re‑analysed, the publicly available data as to the link between the consumption of Vioxx and the occurrence of cardiovascular disease. His conclusion was:
My consideration of all the publicly‑available evidence relating to the principles of causality … and considering how data accumulated over time, leads me to conclude that the weight of the evidence at all times between March 2000 (when the results from VIGOR were released) and September 2004 (when Vioxx was withdrawn) favoured a causal explanation. I conclude that Vioxx is likely to have caused excess [cardiovascular disease], most particularly [myocardial infarction].
Prof Madigan also studied the available data, but with an emphasis on the studies and trials that had been conducted by Merck. He concluded:
I conclude that Vioxx is causally associated with an increased risk of an array of cardiovascular thrombotic adverse events. The statistical evidence is strong and consistent in the major blocks of trials I examined. Even in the short duration, relatively small [osteoarthritis/rheumatoid arthritis] studies the relative risk estimates and p‑values are strongly indicative of a causative association.
308 Prof Cleland held a similar view. He was, from an early date, aware of the FitzGerald hypothesis, and the results of the VIGOR trial came as no surprise to him. In his statement he left no doubt but that he strongly believed that the ingestion of Vioxx predisposed towards CVT adverse events.
309 The experts called by the respondents, however, were of a very different mind. Commencing again with the cardiologists, Prof Celermajer examined the published research, and found that it was inconsistent with the existence of the increased risk alleged by the applicant. He also listed a series of problems which, in his perception, were associated with the FitzGerald hypothesis. His conclusion was:
In my view, considering especially the uncertainties outlined concerning proposed mechanisms of harm and the uncertainties outlined concerning interpretation of the clinical data available, the totality of evidence concerning Rofecoxib and acute atherothrombotic CV events is generally not sufficient to establish a “cause and effect” relationship at the level of individual subjects, beyond a reasonable degree of clinical certainty.
Prof Vaughan concluded the presently relevant section of his report as follows:
Taken together, clinical trials are inconsistent as to whether the administration of COX-2 inhibitors alters the risk of cardiovascular events when administered long‑term. There is little evidence of apparent increased risk when COX-2 inhibitors are taken for short duration at the recommended doses … It is my opinion that a knowledgeable practitioner of the art would view the totality of evidence with equipoise, and conclude that the cumulative data fail to establish such a relationship. The factors that contribute to the development of an acute [myocardial infarction] or stroke in a given patient are exceedingly complex and involve multiple known and unknown factors, unrelated to the administration of a COX-2 inhibitor. Accordingly, I would respectfully submit that, based on my expertise and my review of the evidence, the data linking Vioxx with increased cardiovascular risk fail to meet currently accepted, evidence‑based standards of practice. Furthermore, the totality of the data fails to establish causality.
310 Dr Marks responded to the reports of Profs Woodward and Madigan, and undertook his own analysis of the data available to Merck at various times. He concluded that the results of APPROVe arose substantially from the circumstance that the occurrence of CVT events in the placebo arm was uncharacteristically low. He also opined that, in all of the studies in which the occurrence of such events had been shown to be greater than in the comparator arms, the absolute rate was low and in line with normal community experience. His conclusion was:
From my comprehensive review of Vioxx RCT results and of the expert reports by Professors Madigan and Woodward, I do not find evidence to support applicant claims that Vioxx is pro‑thrombotic.
311 I shall commence by identifying the extent of agreement amongst the experts. In preparation for their concurrent evidence session, the cardiologists were asked a number of questions, including the following:
On the basis of current scientific knowledge, does Vioxx increase a person’s risk of the [pleaded cardiovascular conditions] above what it would have been had the person been:
(a) not taking any medication for arthritis instead of Vioxx;
(b) taking other COX-2 selective inhibitors;
(c) taking a non‑naproxen NSAID instead of Vioxx;
(d) taking naproxen instead of Vioxx,
in otherwise identical circumstances?
312 The cardiologists addressed first the risk of myocardial infarction. They dealt with the first part of the question (where the comparator was taking no medication at all) as though it related to the applicant himself, rather than to persons in general. Profs Zipes and Harper were of the opinion that, compared with taking no medication, the consumption of Vioxx increased the applicant’s risk of having a heart attack by a factor of approximately two (the view of Prof Harper) or of at least two (Prof Zipes). Profs Celermajer and Vaughan said that it was difficult to imagine that the applicant could have had no treatment at all for his arthritis, and that it was, therefore, difficult to assess his risk of suffering a myocardial infarction in the presence of untreated arthritis. They noted that there were two studies which suggested the possibility of an increased risk of a myocardial infarction when Vioxx was consumed in the recommended doses, compared to the consumption of a placebo. Those studies were APPROVe and that published as Kearney (2006).
313 The cardiologists dealt with the remaining parts of this question as they were framed, namely, as relating to persons in general. As to the second part (where the comparators were other COX-2 inhibitors), Profs Zipes and Harper noted that the only other COX-2 selective inhibitor available in Australia at the time when the applicant was taking Vioxx was Celebrex. The standard dose of Celebrex in Australia at that time was 200 mg whereas a standard dose of Vioxx was 25 mg. Profs Zipes and Harper expressed the view that the relative risk for heart attack between Vioxx at that dosage and Celebrex was approximately 1.5 (Graham 2005). Based on several epidemiological studies and meta‑analyses, they opined that Vioxx had the greatest relative risk for cardiovascular events, followed by other COX-2 inhibitors, in an order reflecting their degree of COX-2 inhibition. Profs Celermajer and Vaughan considered that there were insufficient high quality data comparing the cardiovascular outcomes as between different COX-2 inhibitors. They believed that the most reliable estimate came from Kearney (2006), which suggested that the cardiovascular risk of taking Vioxx at a rate of 25 mg daily did not significantly differ from that arising from the consumption of other COX-2 inhibitors. They pointed out that the data in Kearney (2006) were derived from randomised trials, rather than being observational data, as was the case with Graham (2005).
314 As to the third part of this question (where the comparator was a non‑naproxen NSAID), the cardiologists pointed out that there were no randomised controlled data with respect to this comparison. As to such data as there were, Profs Zipes and Harper proffered the interpretation that all NSAIDs (with the exception of naproxen) increased the risk of adverse cardiovascular events, but to a lesser degree than Vioxx. The variation between the data in the various studies highlighted the difficulties and limitations in the interpretation of meta‑analyses and observational data. They considered that the most reliable data came from randomised double blind placebo‑controlled studies. Profs Celermajer and Vaughan took the view that the cumulative data showed similar rates of cardiovascular risk when Vioxx was compared with non‑naproxen NSAIDs, relying on Chen (2007) and on Merck’s pooled analyses of the data from its arthritis and Alzheimer’s disease studies.
315 As to the fourth part of this question (where the comparator was naproxen), Profs Zipes and Harper said that, although the data were variable, and obtained from meta‑analyses and observational studies, the results suggested, at best, that naproxen had a small cardiovascular protective effect, in the order of 10% or less, in which respect they relied on the report of Prof Woodward, to which I shall come presently. Therefore, according to them, taking Vioxx instead of naproxen would increase the risk of an adverse cardiovascular event by a factor of a little more than two. Profs Celermajer and Vaughan considered that the effect of naproxen on cardiovascular risk most likely depended on whether it was taken regularly: if so, it would have an antiplatelet effect similar to that of aspirin (eg Weir 2003, Capone 2004). They said that this effect may have contributed importantly to the result of the VIGOR study. They noted that observational studies suggested a much smaller protective effect, but they considered that this was potentially due to less frequent usage (in part because of gastrointestinal intolerance). They said that other randomised controlled trials in which naproxen was compared with a different coxib, suggested that it had cardiovascular effects similar to those which, in their view, were seen in VIGOR.
316 In their joint report, the cardiologists then turned to the other pleaded cardiovascular conditions (ie other than myocardial infarction). With respect to thrombotic stroke, Profs Zipes and Harper said that they had not undertaken a detailed examination of the data linking Vioxx with this condition. They considered that it was important to note that thrombotic stroke consisted both of atherothrombotic and of non‑atherothrombotic types of ischaemic stroke. Profs Celermajer and Vaughan said that there were no data linking the consumption of Vioxx to thrombotic stroke. To the contrary, they pointed to what they described as “compelling data to show that Vioxx does not predispose to thrombotic stroke”. They further noted the stroke data tabulated in Dr Marks’ Appendix B to the biostatistician’s joint report, which set out the results from various studies of Vioxx conducted by Merck.
317 With respect to unstable angina, transient ischaemic attack and peripheral vascular disease, the cardiologists said that there were insufficient data to draw meaningful conclusions concerning any Vioxx‑related risk.
318 As will be apparent from the foregoing, in forming their views on the question of risk, the cardiologists relied heavily on the published results of various trials and studies. Given that the mechanistic explanation of the relationship between the consumption of Vioxx and cardiovascular risk was, at all times while Vioxx was on the market, a matter of hypothesis, that reliance was inevitable. That naturally takes one to the evidence of the biostatisticians, whose discipline is concerned with the extent to which useful general conclusions as to associations may be drawn from the data yielded by individual, and collective, studies.
319 The questions upon which the biostatisticians were asked to report were grouped by reference to data available to Merck (i) down to December 2003; (ii) down to September 2004 and (iii) as at October 2004 and thereafter. In each case they were asked whether –
… the data from the randomised clinical trials available to Merck … show[ed] a statistically significant difference in the risk of the [pleaded cardiovascular conditions] for Vioxx relative to:
(a) non‑naproxen NSAIDs;
(b) naproxen;
(c) placebo.
As at each of the first two mentioned dates, the biostatisticians were asked whether “the data available to Merck suggest[ed] a real (not fanciful) possibility that Vioxx was associated with an increased risk” of the pleaded cardiovascular conditions. With respect to October 2004 and thereafter, they were asked whether the data established that the consumption of Vioxx increased the risk of the pleaded cardiovascular conditions. It is this final aspect of the matter with which I am presently concerned, although, as it happened, each of the biostatisticians would have answered that question the same way as he would have answered the two earlier parts.
320 Of the pleaded cardiovascular conditions, “due to the lack of data on the other endpoints”, the biostatisticians were able to answer the questions posed of them only in respect of myocardial infarction. They were also able to comment on the CVT endpoint, but it must be appreciated that that dimension of their evidence related to a basket of conditions rather than to any one event. They interpreted the present question as if asked whether Vioxx was “a likely causation” of myocardial infarction and of CVT events generally. Profs Madigan and Woodward answered the question in the affirmative, while Dr Marks answered it in the negative. That is to say, Profs Madigan and Woodward gave the opinion that Vioxx most likely caused myocardial infarction and CVT events, while Dr Marks gave the opinion that it did not. Their joint report did not contain any direct elaboration upon these conclusions: their views were to be found in their respective individual reports.
The Bradford Hill criteria
321 Towards the end of his written submissions on the matter of risk, the applicant referred to, and relied on, a list of (originally nine) criteria proposed in 1965 by Sir Austin Bradford Hill (Hill 1965), by reference to which questions of causation in epidemiology might conveniently be addressed. Prof Woodward said that the criteria had since been adapted to produce the following seven points:
1) There should be evidence of a strong association between the risk factor and the disease. Weak relationships may be due to chance occurrence and are more likely to be explainable by confounding.
2) There should be evidence that exposure to the risk factor preceded the onset of disease.
3) There should be a plausible biological explanation.
4) The association should be supported by other investigations in different study settings. This is to protect against chance findings and bias caused by a particular choice of study population or study design.
5) There should be evidence of reversibility of the effect. That is, if the “cause” is removed the “effect” should also disappear, or at least be less likely.
6) There should be evidence of a dose‑response effect. That is, the greater the amount of exposure to the risk factor, the greater the chance of disease.
7) There should be no convincing alternative explanation. For instance, the association should not be explainable by confounding.
Prof Woodward stressed that none of the criteria was either necessary or sufficient to establish causality, but that each would strengthen the evidence towards such a conclusion. Prof Woodward and Prof Celermajer agreed that the criteria were not a hard and fast set of rules (or, as Prof Woodward put it, that they did not require merely the “ticking of boxes”). Rather, they were points which (in Prof Celermajer’s words) could be “useful in trying to assess a body of evidence as regards a potential cause for certain observations”, or (in Prof Woodward’s words) constituted “a widely‑understood general guide for weighing the evidence of causality”.
322 It is important to keep in mind the original purpose of these criteria. It was to help bridge the gap between association and causation. As Hill himself said in his address to the Royal Society in 1965:
Our observations reveal an association between two variables, perfectly clear‑cut and beyond what we would care to attribute to the play of chance. What aspects of that association should we especially consider before deciding that the most likely interpretation of it is causation?
In his report, Dr Marks captured this sense of the criteria when he said:
No single analysis within a study leading to statistical significance and, in fact, no single study showing multiple statistically significant results is generally accepted as proof of causation. It is not unusual, and even expected, that statistical significance will result simply by chance from performing numerous statistical outcome comparisons within a study and across multiple studies. Statistical significance and causation are substantially different concepts. … Proof of causation only results when well accepted criteria, such as those established by Bradford Hill (1965), are met after reviewing all available relevant scientific information.
In the application of the criteria, in Dr Marks’ view, one should look first at the first, fourth and sixth items, and, if evidence of causation is apparent at that level, one should then move to consider the remaining criteria.
323 There was some minor disagreement between Prof Woodward and Prof Celermajer as to detailed aspects of the Bradford Hill criteria. In his original report, Prof Celermajer identified the first, third, fourth and sixth of these criteria, together with an additional one, “specificity” (giving the example of being run over by a truck as one in which the resulting injuries would be specific to the presumptive cause). Prof Woodward considered that Prof Celermajer was wrong to omit the principle of temporality (the second on the list) which he (Prof Woodward) described as “the most important principle of causality”. He considered that Prof Celermajer was also wrong to omit the principle of lack of an alternative explanation, the seventh on the list. He said that Prof Celermajer was being “too prescriptive” by including the principle of specificity, one of Hill’s original criteria but, according to Prof Woodward, no longer commonly employed. He said that the specificity requirement – that the cause leads to a single effect rather than to multiple effects – clearly had no application in the case of smoking, which was a well‑known risk factor both for cardiovascular disease and for lung cancer. In his statement in reply, Prof Celermajer criticised the conclusions drawn by Prof Woodward by reference to the criteria on his list, but ventured no criticism of the content of the list as such.
324 It was not suggested by either side – or by any of the experts – that the Bradford Hill criteria did not provide an appropriate framework by reference to which questions of risk arising in the present proceeding might be addressed. The applicant stressed, and I accept, that the existence of a risk of a certain kind and degree was a question of fact which, in a court, had to be decided on a balance of probabilities by reference to all of the available evidence. However, as I have said, he relied on these criteria, as did the respondents. For the most part (and to the extent that it was otherwise I shall deal with it), the major themes of the parties’ cases on risk can be accommodated within these criteria. Those themes were (i) the strength of the observed association between the consumption of Vioxx and CVT events including myocardial infarction; (ii) the consistency of that association (including issues of generalisability); (iii) the presence or absence of a convincing alternative explanation for the association observed; and (iv) the presence or absence of a plausible biological explanation. Less conspicuous themes were temporality, reversibility and dose‑relationship. Each of these can be accommodated within the Bradford Hill criteria.
325 The focus of the epidemiological debate before the court was on aspects relevant to the first and fourth of the Bradford Hill criteria – strength and consistency of association. The applicant submitted that the association between the consumption of Vioxx and CVT events, particularly myocardial infarction, was strong and consistent as between study settings. The respondents could not deny that there were studies which had revealed a strong association, at least in relation to myocardial infarction, but they submitted that there were reasons for this that stood in the way of reaching any general conclusion as to causation or risk, and that the evidence of a like association across different study settings was patchy and inconsistent.
326 The applicant relied primarily on the results of the APPROVe trial, because that was the first placebo‑controlled prospective RCT in which the reported excess of CVT events in the Vioxx arm was statistically significant. The only previous RCT to have shown such a statistically significantly excess was VIGOR, but there the comparator was naproxen, not placebo. Some (but not all) other trials and studies had shown an excess of events in the Vioxx arms, but not at a statistically significant level. As Prof Harper put it, “… the APPROVe result … is scientifically sound. Everything else has limitations.”
APPROVe
327 As stated elsewhere in these reasons, a result of the APPROVe trial, as reported to the FDA and as published as Bresalier (2005), was that the relative risk of encountering a CVT event (as defined in the CV SOP) from taking Vioxx rather than placebo was 1.92 (1.19, 3.11). The corresponding figure for the APTC endpoint was 2.06 (1.16, 3.64). For myocardial infarction, the relative risk was 2.53 (1.11, 6.28) and for all cardiac events it was 2.80 (1.44, 5.45). Based on these statistically significant results, Profs Zipes and Harper expressed the opinion that the consumption of Vioxx about doubled the risk of the occurrence of a CVT event. Prof Harper pointed particularly to the relative risk figures for cardiac events and myocardial infarction in support of the opinion which he expressed. He said:
[T]he APPROVe study to me is the hallmark, is the gold standard. I think the VIGOR study, we are talking about interpretations in 2000 but if you interpret the VIGOR study today with the knowledge we have, with the additional knowledge we have, I think that’s the second best evidence regarding the risk of Vioxx and heart attack. I think all the other studies, you can pick about – there are about 10 studies and you can pick whatever result you like, almost, out of the meta‑analysis and observation studies. I think that data is much weaker. But as Professor Zipes has said, if you take it in totality, it generally … shows an increased risk with Vioxx. So I would say that the APPROVe study is … certainly the gold standard that we relied on to say that we think the risk – [the applicant’s] risk was increased by a factor of 2, and there has been a follow‑up to the APPROVe study, just following patients for longer, which again … reinforce … the original results of the APPROVe study.
328 The follow‑up analysis referred to by Prof Harper was Baron (2008). I shall refer to it below. It was submitted on behalf of the applicant that the studies mentioned by Prof Harper amply justified the conclusion that the consumption of Vioxx increased the risk of suffering the pleaded cardiovascular conditions.
329 The respondents ventured no criticism of the APPROVe trial, nor of the conclusions reached in it. Profs Celermajer and Vaughan described it as “a good study”. However, the respondents submitted that there were four “key limits” on the uses for which the results of APPROVe might be deployed:
(a) APPROVe does not sit “in splendid isolation”. It is necessary to consider the totality of the evidence properly to understand the results. A large body of data exists that do not support a cardiovascular risk with Vioxx. The data are conflicting and inconsistent.
(b) The cardiovascular risk hazard curves reveal that the risk is only apparent after 18 months. Therefore, the data from APPROVe itself are confusing.
(c) In the absence of an established mechanism, the data can only be considered at the population level. The data cannot be applied to an individual.
(d) APPROVe was tested on patients with colon adenomas, whose “cardiovascular risk profile” differs from that of the general population. Therefore, the APPROVe data cannot be readily applied to the general population with any meaningful results.
330 The first of these points effectively calls up the fourth Bradford Hill criterion. It is best considered in conjunction with the applicant’s submission that the APPROVe results derive support from the results of other trials and studies when properly understood. Indeed, the applicant made no suggestion that APPROVe sat “in splendid isolation”: quite the contrary. The third of the respondents’ points effectively calls up the third of the Bradford Hill criteria – the existence of a plausible biological explanation – but it raises also the question whether the existence of a risk at the population level justifies a conclusion of causation in the case of a particular individual. I shall return to those questions. The fourth point may raise questions under the fourth and the seventh of the Bradford Hill criteria; or it may sit outside them altogether, in the sense that it seems to propose that, even if it might be concluded that Vioxx caused CVT disease in patients with colon adenomas, it did not follow that it did so in other patients. I shall return to these issues also. The second point concerns the way the APPROVe results themselves should be interpreted, and it is to it that I next turn.
331 The point concerns the timing of the occurrence of CVT events in the respective arms of the APPROVe trial. A Kaplan‑Meier plot graphs the cumulative percentage of patients with events over time. The cardiovascular safety report for APPROVe set out the following Kaplan‑Meier plot for the two arms, Vioxx and placebo:
The interpretation of this graph given in the report was:
The curve for the rofecoxib treatment group appeared generally linear from 0 to 36 months while the curve for the placebo treatment group appeared generally linear during the first 18 months and then the curve flattened during 18 to 36 months. The two curves appeared to be similar during the first 18 months and then started to separate at approximately 18 months.
According to a table in the report, over the first 18 months, the relative risk of suffering a thrombotic cardiovascular event from taking Vioxx was 1.18 (0.64, 2.15). Over the balance of the period, the figure was 4.45 (1.77, 13.32).
332 The authors of the report went further and said of this apparent divergence at 18 months:
Figure 3 and Tables 6 and 7 demonstrate that the hazard ratio or the relative risk for thrombotic cardiovascular events did not appear to be a constant over time. This was confirmed by the testing of the proportional hazard assumption for the Cox proportional hazards model (p=0.014).
A similar conclusion was expressed in the published article setting out the results of APPROVe, written by the members of the Steering Committee and two members of the External Safety Monitoring Board for the trial: Bresalier (2005). That aspect of the article led to some scientific controversy, and to the publication of a correction by the authors in the New England Journal of Medicine in July 2006. In that correction, the authors stated that they had originally used linear time rather than the logarithm of time as specified in the Methods section of their article, in the test for proportionality of hazards. They withdrew the statement that “[t]he changing pattern of the treatment effect over time was confirmed by a failed test for proportionality of hazards (P = 0.01)”, and replaced it with a statement that “the results of an overall test of the proportional‑hazards assumption for the entire 36‑month observation period did not reach statistical significance (P = 0.07).”
333 The 18‑month aspect of the APPROVe results was emphasized by Dr Marks and Prof Celermajer in their respective original reports. Dr Marks said: “A visual inspection of the results … clearly indicates a pattern of overlapping Vioxx and placebo curves through 18 months, after which a clear separation occurs.” He pointed out that, in both arms, the average age of the patients was 59 years at the commencement of the trial. After 18 months, the average age would have been 60.5 years. Dr Marks opined that it would not be surprising to observe a slight increase in the rate of occurrence of adverse events in the later part of the trial, reflecting the increased risk of cardiovascular events which comes with older age. Such a slight increase was indeed observed in the Vioxx arm. However, the major contributor to the separation in the results after 18 months was a reduction in the rate of occurrence in the placebo arm.
334 While noting the technical complications involved in relying on the visual appearance of the Kaplan‑Meier plot originally published in Bresalier (2005), Prof Celermajer proposed that the delayed separation of the two curves was inconsistent with an early occurrence of pro‑thrombotic events, which would be expected if the FitzGerald hypothesis were valid. This was one of the main pieces of evidence that gave Prof Celermajer cause to pause before believing that the hypothesis might be valid. However, in the concurrent evidence session Prof Celermajer accepted that the Kaplan‑Meier curves did not give one a basis for saying that the effect of Vioxx emerged only after 18 months. He said:
[A] visual inspection of the CV risk hazard curves … suggests that these curves are broadly similar for the first 18 months and diverge after this point, up to the 36 month study point. Statistical analysis of these hazard curves, however, do not suggest that the risk difference between groups progresses differently over time … The study was designed for a 36 month follow‑up period and it is clear that, at the 36 month time point, Rofecoxib was associated with a statistically significant increased risk of serious CV events, as prospectively defined.
Prof Vaughan said:
[If] you look at the data, and people look at it in all different ways, the rates of myocardial infarction in the Vioxx treated group and the placebo treated group track – they overlap, pretty much, for the first year and a half, for 18 months, and the rates are very similar and what happens after 18 months is the rate continues at the same level for Vioxx and it drops off for the placebo group. Now, I don’t doubt that those numbers are real but I don’t understand why all of a sudden the placebo group would drop off in terms of their rates of events. So it’s a peculiarity. It may be a red herring but it’s an oddity of that study.
335 If we move from the visually‑apparent separation of the curves in the Kaplan‑Meier plot at 18 months to the technical means by which the statisticians would measure whether there was any significant difference in the proportional rate of occurrence of events in two arms of the study over time, we come to what Prof Madigan described as the “proportional hazards” assumption. He explained it in the following terms:
The “proportional hazards” assumption plays an important role in survival analysis and greatly simplifies many analyses. For confirmed CVT events in APPROVe, it amounts to assuming that the ratio of the “hazard function” for a Vioxx patient to the “hazard function” for a placebo patient does not vary over time. The hazard function is a‑function of time and essentially represents the probability of having a first event at a given moment. So if Vioxx patients have a hazard function that is, say, double that of placebo patients, the proportionality assumption says that this doubling stays constant over time. The assumption presumably is never perfectly true but the general idea is that it is often close enough to being true that it represents a reasonable basis for analysis.
It was in relation to this test that Bresalier and colleagues made the mistake to which I have referred above. As to that mistake, Dr Marks said:
The original proportionality test reported in the Bresalier publication produced a p value of p < .01, indicating rejection of the hypothesis of proportionality, and leading to a conclusion that the CVT event curves differed for the VIOXX and placebo drug arms. The issue arose when Merck discovered that the proportionality test they computed and reported was different than the prespecified test. Merck then reran the analysis using the specified statistical test for proportionality, which resulted in p < .08.
Dr Marks said that “both proportionality tests are correct, but are based on different statistical assumptions.” He said that it was important to detect when interactions occur in clinical research –
… so a well established statistical rule for a pre‑specified interaction test is to use a more liberal cut point for determining statistical significance, at least p < .10, and more commonly p < .15 or even p < .20. … Using any of these alternative and acceptable cut points for the APPROVe pre-specified proportionality analysis would lead to the same conclusion for either of the two Merck reported proportionality tests. The appropriate conclusion from Merck’s proportionality analysis (both test results) is that there is statistical evidence of non-proportionality.
336 The applicant submitted that what he described as the “18 month theory” had been “debunked” by Dr SW Lagakos (2006). The applicant correctly observed that Lagakos’ conclusion was that “one could not conclude from the data that a shorter course of rofecoxib is safe”. Prof Madigan explained Dr Lagakos’ analysis as follows:
As Lagakos … points out, a claim that the cumulative incidence rates were identical in the first 18 months neither was demonstrated by nor follows from the use of either of the interaction models used to test the proportional hazards assumption. Put another way: even if there did exist unequivocal evidence against proportionality, this would not imply the existence of an 18‑month changepoint per se; many different kinds of departure from proportionality could produce a significant test.
Or, as Prof Celermajer put it, Lagakos drew the conclusion that it was not possible to make any conclusion one way or the other about proportionality from the study.
337 Prof Zipes pointed out that Merck had not pre‑specified the possibility of a change in the relative cardiovascular toxicity of Vioxx after 18 months of the trial (or after any other particular period), and that its treatment of the results was, in this respect “post hoc” and, therefore, statistically questionable. He also said that, if account were taken of all investigator‑reported CVT events, the separation between the rates of occurrence in the two arms appeared much earlier than the 18‑month point. He added that a “subsequent article stated that the calculation was wrong and that there was earlier separation of the curves.” Prof Harper referred to the subsequent academic criticism of the statistical conclusion originally expressed by Bresalier (2005).
338 The applicant relied also upon the follow‑up data published, and discussed, by Baron (2008). He submitted that that study had “confirmed a near immediate onset of increased risk from Vioxx in APPROVe”. However, the conclusion of Baron and colleagues as to the timing of the emergence of an excess of events in the Vioxx arm related to the APTC endpoints, not to CVT events. They said:
The APPROVe protocol during the active treatment phase of the study included toxicity follow‑up of patients during treatment and the following 14 days. However, by censoring follow‑up off treatment, this approach might overlook adverse events that are not temporally linked to use of rofecoxib but nonetheless associated with the drug. Therefore, after cardiovascular toxic effects became apparent, the study protocol was amended to include follow‑up of all randomised participants for at least 1 year off treatment. The previous APPROVe cardiovascular report was criticised for the absence of data from such follow‑up. Furthermore, some questioned the statistical analysis of the timing of the increased risk, disagreeing with the analytic approach used and noting an acknowledged error in the analysis report. The observation that the increased risk linked to the treatment with rofecoxib emerged only 18 months after initiation of treatment was especially controversial.
Because of small numbers, the authors were unable to present the Kaplan‑Meier analysis for CVT events, but they did conclude that their data were “compatible with an early increase in risk that seems to persist for about 1 year after 3 years of treatment”. They did, however, have sufficient data to prepare a Kaplan‑Meier plot for APTC events. It was as follows:
The authors stated:
Investigation of the time course of the raised risk is hampered by small numbers of events. However, the HR [hazard ratio = relative risk] for the APTC endpoint did not substantially change over time, and our data are compatible with an early increase in risk for these events.
They also stated that, although their subgroup analyses suggested that individuals with risk factors for cardiovascular disease had higher rofecoxib relative risks than did healthy individuals, their data were not conclusive on that point.
339 Notwithstanding the inability of the authors in Baron (2008) to construct a Kaplan‑Meier plot for CVT events, Merck itself appears to have done so in 2006. In an analysis dated 26 May 2006 referred to (in another respect) in the evidence of Dr Marks, and to which I have referred in paras 195‑196 above, Merck presented such a plot of the occurrence of CVT events in the APPROVe study. They presented the data in two presently relevant ways. First, they included all events that had occurred up to the 210th week of study therapy for the patient concerned. The plot was as follows:
Secondly, and as an alternative way of looking at the relative emergence of events, they included all events that had occurred up to 31 October 2005. The plot was as follows:
The two Kaplan‑Meier plots were in the evidence, but were not referred to by any party. Neither were they referred to by any witness. The document in question was tendered because it had been referred by Dr Marks for other purposes. Under cross‑examination, he was not taken to these plots, notwithstanding that they appeared in a document on which he relied. I do not know what he, or the other biostatisticians, would have made of them. Neither do I know what to make of the statement in Baron (2008) that they were unable to analyse CVT events by way of a Kaplan‑Meier plot.
340 What did the respondents make of this 18‑month aspect of the APPROVe results? As I have said, in their written submissions, they said that the data were “confusing”. They referred to the evidence of Dr Marks that there were instances in medical research where the placebo arm of a study “outperforms” the active arm. They referred to the agreement of the biostatisticians that there was “some evidence of non‑proportional hazards in the APPROVe study”. They referred to the evidence of Profs Celermajer and Vaughan that, if the FitzGerald hypothesis were valid, one would have expected to see a higher relative rate of thrombotic events in the Vioxx arm of the trial well within the first 18 months. However, the respondents’ written submissions left this aspect of the debate hanging there, as it were, and their oral submissions took the matter no further. There was no explanation as to how or why (if indeed it was the respondents’ case) the delayed appearance of the increased relative rate of occurrences in the Vioxx arm of APPROVe required some kind of qualification to the general conclusion that the consumption of Vioxx was associated with an elevated risk of CVT events.
341 On this state of the evidence and the submissions, I do not think I am entitled to find, favourably to the respondents, that the results of the APPROVe trial justify the conclusion that the true effect of Vioxx with respect to CVT events became apparent only after about 18 months of medication. I accept Dr Marks’ evidence (which was not challenged) that proportionality was rejected where p < 0.08, but this was apparently regarded by Bresalier and colleagues as falling short of statistical significance. Prof Celermajer also so regarded it. Another circumstance, as Prof Zipes pointed out, is that APPROVe was not designed to test for proportionality over time, and that the observation made by Dr Marks, while legitimate as such, was “post hoc”. The respondents themselves have described the results as “confusing”, and Prof Vaughan thought that the falling away of the events in the placebo arm was “a peculiarity” and “an oddity”. With respect, I can well understand why the respondents and Prof Vaughan would have taken that position. It does seem rather odd that the true pattern of CVT events in a placebo situation – ie in a situation in which the patient is taking nothing at all – would be such that, in any 36‑month period, more such events would occur (otherwise than by chance) in the first 18 months than in the second 18 months. Looking at the matter from the purely biostatistical perspective, if otherwise it is proper to conclude that the APPROVe trial showed that Vioxx was associated with an increased risk of CVT events, I am not disposed to qualify that conclusion by reason of the apparent divergence in the rates of occurrence in the two arms after 18 months.
342 The APPROVe trial did show such an association. At the 95% probability level, it established (at least for a population of persons having the characteristics there studied) that the true relative risk of encountering a CVT event from taking Vioxx, in comparison with taking nothing at all, fell within the range of 1.19 – 3.11. For myocardial infarction, the range was 1.11 – 6.28. For forensic purposes, I am bound to regard this as an association of apparent strength.
The generalisability of the APPROVe results
343 It is convenient next to consider the respondents’ submission that there is, or at least may well be, something about patients with colon adenomas that make them atypical of the population as a whole with respect to the risk of cardiovascular disease. In his witness statement, Prof Celermajer made a deal of this point. He referred to a randomised double‑blind trial of aspirin (against placebo) as a preventative agent against colorectal adenomas (Baron 2003). Although statistically non‑significant (except in the case of high‑dose aspirin, where the relative risk (compared to placebo) of a CVT event was 10.0 (1.29, 77.73)), there was an apparent positive (and dose‑dependant) association between taking aspirin and the occurrence of CVT events, including myocardial infarction. Prof Celermajer said that the findings of this trial appeared “quite strikingly discordant with the wealth of information available about the [beneficial] effects of aspirin on [cardiovascular] event rates in very large primary prevention trials.” He referred also to a study of the effectiveness of celecoxib for the prevention of colorectal adenomas published as Solomon (2005) and known as the “APC” (Adenoma Prevention with Celecoxib) study. That was a placebo controlled RCT involving doses of celecoxib of 200 mg and 400 mg twice daily, in which it was found that the relative risk of encountering a cardiovascular endpoint (including death) from taking celecoxib 400 mg rather than placebo was 3.4 (1.4, 7.8). This result was out of conformity with the results of studies of celecoxib involving other patient groups (White 2003; ADAPT 2006). It was, however, difficult to reconcile with a similar study to assess the recurrence of colorectal adenomas in patients who had had polyps of the large intestine (the “pre‑SAP” trial), where a moderate non‑significant excess of serious CVT events was shown amongst patients taking celecoxib rather than placebo (Arber 2006). Prof Celermajer also referred to a study which combined the APC data with the pre‑SAP data, and which reached the conclusion that there was “a nearly twofold” increased cardiovascular risk in the colon adenoma population taken as a whole (Solomon 2006).
344 Although accepting that there were limitations in these celecoxib data, Prof Celermajer noted that they were consistent with the results of two other analyses. The first was an analysis of the results of trials of coxibs in colon adenoma patients and arthritis patients (Scott 2007). The point estimate for the relative risk of taking coxibs was 2.67 in colon adenoma patients and 1.6 in arthritis patients. The second analysis compared the occurrence of vascular events in trials of coxibs lasting a year or more, in which it was found that the relative risk of taking coxibs was 1.80 in the case of colon polyp patients and 1.14 in the case of Alzheimer’s disease patients (Kearney 2006). Prof Celermajer then considered whether there may be a mechanistic explanation for the apparent susceptibility of the colon polyp group to CVT disease, but such possibilities as presented themselves were considered to be “speculative”. His conclusions included the following:
In summary, there are data to suggest that patients with colon adenomas may have a [cardiovascular] response to COX inhibition (non‑selective or selective) that may be different to the general population. … I must re‑emphasise that the APPROVe data certainly stand alone, however, as evidence of a significant association between Rofecoxib and excess [cardiovascular] risk in the population studied. As noted above, however, there is a considerable difference in the “burden of proof” required for a drug to be suspected of a safety concern and thus subject to a market withdrawal versus the burden of proof required to prove causation for the general population, with scientific certainty.
In the context of the latter and like observations, in his oral evidence Prof Celermajer made it clear that, by “scientific certainty”, he had in mind a conclusion that could be reached with a 95% probability of being valid.
345 Although both Prof Harper and Prof Zipes disagreed with Prof Celermajer on these issues, they did not engage with his argument at the level of detail. Prof Harper did not accept that the risk of encountering a CVT event as a result of taking Vioxx might be qualified, or somehow calibrated, by reference to some non‑vascular condition from which a particular patient was suffering. He said:
I think the relevant point is whether a person has atherosclerosis more than anything else. It doesn’t matter whether they have got osteoarthritis or other condition. If they have – I think coronary atherosclerosis is a necessary substrate for the adverse action of Vioxx to occur and many people, of course, have clinically silent atherosclerosis, they don’t know that they have got it. … I think that’s the important issue, not whether [the applicant] had osteoarthritis or ankylosing spondylitis or any other medical condition and I think if you analyse the data with Vioxx, the risk of an adverse cardiovascular event is greater in people that are either known to have atherosclerosis or have the risk factors that make it likely that they have clinically silent atherosclerosis.
In his statement in reply to Prof Celermajer, Prof Zipes said that it was hard to conceive of a more diverse population of patients than the rheumatoid arthritis group (in VIGOR), the colonic polyp group (in APPROVe) and the Alzheimer’s group, in each of which, in Prof Zipes’ assessment, Vioxx had been shown to cause harm. To those groups he would add patients with osteoarthritis, “yet a fourth disease entity”. In his oral evidence, he said:
I think the evidence or the scientific data which I have reviewed shows an increased cardiovascular risk with Vioxx in diverse populations, osteoarthritis, rheumatoid arthritis, patients with adenomas and I see Alzheimer’s increased in death and I see nothing that would say the risk is specific to a single population of patients. As a matter of fact, the data are more convincing of [the lack of] a heterogenous effect than many other scientific trials that I have looked at or participated in where there is a much narrower population study.
346 In his oral evidence, Prof Celermajer made it clear that he did not take the position that the results of APPROVe were not generalisable because the trial dealt with patients with a particular condition. Rather, his position was that, “if one were asked the question, ‘Is it generalisable?’ one would look to the evidence and see some uncertainty on that point .…” He agreed that APPROVe was part of Protocol 203, and that the court was entitled to understand that Merck at least thought APPROVe was an appropriate element of that protocol for the purpose of investigating the cardiovascular risks, if any, associated with Vioxx. He agreed also that APPROVe was the best study relevant to the question with which the court was concerned.
347 Prof Celermajer’s opinion that there was “some uncertainty” as to the generalisability of the APPROVe results was based on a measured and, if I may say so with respect, credible consideration of the results of such other studies as had the potential to throw light on the subject. He was not challenged on that work at the level of detail, either by the applicant or by the cardiologists whom he called. The latter dealt with the question of generalisability at a high level only. It is self-evident that Merck considered APPROVe to be an appropriate element of the protocol drafted for the purpose of investigating the cardiovascular risks associated with the consumption of Vioxx. But it was only one element. The point of studying cohorts of patients with different conditions was, conformably with the third Bradford Hill criterion, to assess consistency of results in different study settings. The quality of APPROVe as a study is, of course, a different matter from that of generalisability. I am, in the circumstances, prepared to accept that the APPROVe results reflect, at the 95% probability level, the true relative risk of taking Vioxx rather than placebo amongst persons in the broader population who have the characteristics of the patients in the trial. But I also accept the evidence of Prof Celermajer that there is some uncertainty as to the generalisability of those results to the population at large.
VIGOR and the role of naproxen
348 I turn next to the evidence of other clinical trials upon which the applicant relied. Unsurprisingly, here he placed primary emphasis upon VIGOR. As indicated elsewhere in these reasons, the results of that trial yielded relative risks of taking Vioxx rather than naproxen of 5.00 (1.72, 14.29) in the case of myocardial infarction, of 2.78 (1.35, 5.88) in the case of all cardiac events and of 2.38 (1.39, 4.00) in the case of all CVT events. Principally by reference to the table to which I have referred in para 137 above, Prof Woodward concluded that there was strong evidence of an association between Vioxx and cardiovascular disease, most especially myocardial infarction. In his statement in reply he described this, more accurately in my view, as evidence of a strong association. Prof Celermajer said that the strength of an association was often expressed as the relative risk – “[t]he higher the relative risk, the more likely the exposure is to be causally linked to the effect.” His difficulty with Prof Woodward’s conclusion from VIGOR was that it focussed solely on one event, myocardial infarction. In this respect, Prof Celermajer relied upon the following passage in his original report:
In my view, what would concern a patient (“a priori”) if there were a question of [cardiovascular] risk, would be any serious [cardiovascular] event, which would include cardiac death, [myocardial infarction] and stroke; I do not think that a patient would prospectively be worried about [myocardial infarction] to the exclusion of stroke, for example, especially as the pathogenesis of these events is closely linked (both being complications of atherosclerosis).
Prof Celermajer said that “‘strong’ association means different things to different people and Professor Woodward has made the association in VIGOR appear stronger by focussing on only one (non pre‑specified) endpoint.”
349 In expressing this criticism of Prof Woodward, Prof Celermajer was, in my respectful view, drawing attention to a distinction which is of singular importance in the present case. That distinction is between the risk of suffering one or more of a number of related conditions, on the one hand, and the risk of suffering a particular condition, on the other. I say something more about this distinction elsewhere, but here my comment would be that Prof Celermajer’s criticism of Prof Woodward is too harsh, and may, depending on context, beg the question. It is too harsh because Prof Woodward did express his conclusion with reference to cardiovascular disease generally, albeit that he pointed out, with obvious justification, that the association observed in VIGOR was especially strong when myocardial infarction was the event of interest. It may beg the question in the sense of assuming that the court in the present case is, like the hypothetical patient to whom Prof Celermajer referred, chiefly interested in whether any CVT event would occur as a result of the ingestion of Vioxx. As I explain elsewhere, that is a somewhat simplified perception of the task confronting the court.
350 The controversy surrounding the VIGOR data related not to the apparent strength of association indicated by them, but to the seventh Bradford Hill criterion, namely, whether the circumstance that naproxen was the comparator provided a convincing alternative explanation. As I have indicated above, Merck took the view that the results could and should be explained by the cardioprotective effect of naproxen, rather than by the cardio‑toxic effect of Vioxx. A significant aspect of the applicant’s case in relation to VIGOR was, therefore, the submission that the respondents’ so‑called “naproxen theory” was without substance. Here he relied primarily upon the evidence of Prof Woodward.
351 Prof Woodward undertook a literature search of the studies that had compared naproxen with placebo, and naproxen with other NSAIDs, for risk of myocardial infarction. Typically, the studies derived from large electronic databases of prescriptions or dispensing. He presented the results of his search in the following two-part table:
Table 5.1 Studies of naproxen and MI___________________________
First Pub’n Relative risk
Author date Database Years (95%CI)_______
Direct effects (naproxen v placebo)
Ray 1/02 MEDICAID, 1987-98 0.95 (0.82 - 1.09)
TN
Watson 5/02 GPRD, UK 1988-99 0.61 (0.39 - 0.94)
Solomon 5/02 MEDICAID, 1991-95 0.84 (0.72 - 0.98)
NJ
Schlienger 9/02 GPRD, UK 1992-97 0.68 (0.42 - 1.13)
Raj 10/02 MEDICAID, 1999-01 0.93 (0.82 - 1.06)
TN
Mamdani 2/03 Ontario 1998-01 1.0 (0.6 - 1.7)
Kimmel 3/04 Hospitals, PA 1998-01 0.48 (0.28 - 0.82)
Garcia 6/04 GPRD, UK 1997-00 0.89 (0.64 - 1.24)
Rodriguez-
Graham 2/05 Kaiser Perm, 1999-01 1.14 (1.00 - 1.30)
USA
Levesque 4/05 Quebec 1999-02 1.17 (0.75 - 1.84)
Johnsen 5/05 Denmark 2000-03 1.50 (0.99 - 2.29)
Hippisley- 6/05 QRESEARCH, 2000-04 1.27 (1.01 - 1.60)
Cox UK
_________________________________________________________________________
First Pub’n Relative risk
Author date Database Years (95%CI) Comparator__
Indirect effects (naproxen v other NSAIDs)
Jick 7/00 GPRD, UK 1996-98 1.0 (0.3 - 3.3) Diclofenac
Ray 1/02 MEDICAID, 1987-98 0.83 (0.69 - 0.98) Ibuprofen
TN
Rahme 5/02 Quebec 1992-94 0.79 (0.63 - 0.99) Other NSAIDs
Solomon 5/02 MEDICAID 1991-95 0.82 (0.66 - 0.97) Ibuprofen
USA
Watson 5/02 GPRD, UK 1988-99 0.65 (0.34 - 1.24) Other NSAIDs
Rahme 1/07 Quebec 1999-02 1.16 (0.89 - 1.51) Acetaminophen
352 Prof Woodward pooled the results of these studies on a cumulative basis using random effects meta‑analysis. This enabled him to calculate the pooled relative risk, and the confidence intervals pertaining to that risk, cumulatively at various points in time from the start to the end of the period over which these studies were published. For the placebo comparison, he presented his results in the following figure:
For an overall comparison which covered both placebo and “other NSAIDs”, Prof Woodward presented his results in the following figure:
353 Prof Woodward explained that there were some limitations in the studies to which he had had reference, and some overlapping of the work of those studies. However, making due allowance for those qualifiers, he expressed the view that, for the naproxen/placebo comparison, the lowest point reached by the cumulative best estimate of the relative risk of taking naproxen was 0.84, and the lowest point reached by the lower confidence limit of that risk was 0.72. Taking all the studies into account (ie including both the placebo and the “other NSAIDs” comparisons), the lowest point reached by the cumulative best estimate of the relative risk of taking naproxen was 0.93, and (excepting the result which would have appeared after the first study was published in July 2000) the lowest point reached by the lower confidence limit of that risk was 0.86. In his opinion, the cardioprotective benefit of naproxen implied by these figures was a considerable distance short of providing an explanation for the 5‑fold difference in the risk of myocardial infarction observed as between the Vioxx and naproxen arms in the VIGOR trial.
354 Before expressing his general conclusion, Prof Woodward looked at four published articles which had dealt with the subject of the cardioprotective effect of naproxen. The first was Weir (2003). In that article (which was submitted for publication on 13 May 2002 and accepted on 8 November 2002), the authors referred to four earlier publications of studies which had, amongst other things, examined data which were relevant to this subject. Each of those publications is mentioned in Prof Woodward’s tables, set out above. The first was that by Ray (Jan 2002), in relation to which Weir and colleagues noted that the authors found no association (in the relative rates of hospitalisation for acute myocardial infarction or coronary heart disease death) between the use of any NSAID, naproxen or ibuprofen and the outcome. They (ie Weir et al) said:
The absence of a large protective effect for naproxen could be explained in part by the general population that was represented in the Tennessee Medicaid cohort. Most nonaspirin NSAID use in the community is intermittent – for acute pain and symptoms of osteoarthritis. On the basis of the information that we have previously reviewed, only chronic use of an NSAID with sustained antiplatelet effects would be expected to be cardioprotective.
355 The next previous publication examined by Weir and colleagues was Watson (2002). They referred to the relative risk figure (set out in Prof Woodward’s table above), and pointed out that this figure related to patients who were currently using naproxen. As it happened, the Watson study also contained data in relation to patients who had previously used naproxen, where the relative risk figure was 0.87 (0.65, 1.16).
356 The next publication referred to by Weir and colleagues was Rahme (2002), in which the association between the use of naproxen and the occurrence of acute myocardial infarction had been studied. As is apparent from Prof Woodward’s table, the comparison here was with patients who were using other NSAIDs. The relative risk figures (as set out by Prof Woodward above) related to those who were then currently using naproxen and other NSAIDs. Weir and colleagues added:
This effect seemed only to be present with a concurrent naproxen prescription and was strongest in those who had previously filled at least 2 consecutive 30‑day prescriptions. This is consistent with naproxen’s reversible antiplatelet effects and would suggest that chronic persistent use is required for cardioprotection.
357 Weir and colleagues next referred to Solomon(May 2002), and observed that it had been found that users of NSAIDs generally had the same risk of myocardial infarction as non‑users, but that the use of naproxen was associated with a significant reduction in that risk.
358 Of the previous studies examined by Weir and colleagues, only the first (Ray January 2002) did not disclose, at a statistically significant level, the beneficial effect of naproxen in relation to the occurrence of myocardial infarction. Indeed, to take what might be considered to be the strongest case for the existence of such an effect, in the Watson study the lower limit of the 95% confidence interval for the relative risk of taking naproxen rather than placebo was 0.39. However, Prof Woodward was critical of Weir and colleagues, first, for having made no attempt to quantify the pooled evidence on naproxen (by contrast with having done a pooled analysis of the Vioxx data as such), and secondly, for not having referred to Jick (2000) and Ray (October 2002). He said that the exclusion of those studies from this figure showing the overall results (ie those relating both to placebo and to other NSAIDs) “would cause the pooled evidence to suggest a more extreme cardioprotective effect of naproxen”.
359 Prof Woodward referred to the suggestion by Weir and colleagues that the absence of any apparent naproxen effect in Ray (January 2002) could be due to the intermittent use of naproxen that was typical in the study population there. He (Prof Woodward) said that it was outside his expertise to prove or to refute that suggestion, but that an inspection of the figures in his table (see para 351 above) suggested that the results achieved by Ray and colleagues “were not untypical”. Prof Woodward also expressed the view that Ray and colleagues used a “superior epidemiological design” to that seen in the studies of Rahme, Solomon and Watson.
360 The second published article looked at by Prof Woodward was Juni (2004). The authors of that article undertook a cumulative pooled analysis of the results of previous studies which had examined the relative cardioprotective effect of naproxen, by comparison with placebo and other NSAIDs. Those studies are all captured within Prof Woodward’s list in para 351 above. The most recent was Garcia‑Rodriguez (June 2004), in which the authors found that the cumulative combined relative risk of taking naproxen rather than a comparator was 0.86 (0.75, 0.99). Prof Woodward said that that figure was slightly lower than his own result at the corresponding date, but nothing turns on that minor difference.
361 The third published article looked at by Prof Woodward was Kearney (2006). This study was a meta‑analysis of 138 randomised trials “involving a comparison of a selective COX-2 inhibitor versus placebo or versus a traditional NSAID (or both), in which there were a total of 145,373 participants”. The authors found that the relative risk of suffering cardiovascular disease (not merely myocardial infarction) from taking naproxen rather than placebo was 0.92 (0.67, 1.26). Although the methods and endpoints used by these authors differed from those used by Prof Woodward, he expressed the view that the authors’ pooled estimate of 0.92 was similar to his own.
362 The fourth published article looked at by Prof Woodward was McGettigan (2006). This was also a pooled study of the results of studies published elsewhere. Most of the outcomes were myocardial infarction or sudden death from cardiovascular disease. According to Prof Woodward, the authors found that, compared with non‑use or remote use, the relative risk of using naproxen was 0.97 (0.87, 1.07). Prof Woodward pointed out that this was very close to his own estimate of 0.98. He added that, compared to the use of other NSAIDs, the relative risk of using naproxen was 0.75 (0.63, 0.88), “which confirms the frequently purported hypothesis that naproxen has a lower CVD [cardiovascular disease] risk/greater CVD protection than other non‑selective NSAIDs.”
363 From his own analysis, and using the publications to which I have referred as confirmation, Prof Woodward reached the general conclusion that the consumption of naproxen led to a small reduction in the risk of suffering a myocardial infarction, probably of the order of about 2%. However, Prof Woodward made it clear that his analysis of the naproxen data was a purely statistical one. As mentioned above, he said that it was not within his expertise to prove or to refute the comment by Weir and colleagues that the absence of an apparent naproxen effect in Ray (January 2002) could be due to the intermittent use of naproxen. In his oral evidence, Prof Woodward confirmed that the biological effect of naproxen in a patient, and the significance of compliance in that context, were not statistical questions.
364 From the perspective of a statistician, Dr Marks criticised Prof Woodward’s analysis of the naproxen data. That criticism was not, with respect, substantial. Dr Marks said: “For the reasons I have given above, the Juni and McGettigan publications are not adequate for documenting the true level of naproxen cardioprotection.” But his earlier criticism of Juni related to its treatment of the rofecoxib data, not to its treatment of the pooled naproxen study results. Dr Marks’ only comment about that aspect was:
Juni also provided a meta‑analysis of 11 retrospective studies to evaluate the effect of naproxen on [myocardial infarction]. Juni showed the risk ratio for naproxen was 0.86 (95% CI 0.75‑0.99), and concluded “if a protective effect of naproxen exists, it is probably small”. This result agrees with the RCTs evaluation of naproxen, as summarized in my Figure 9, that naproxen does provide cardioprotection.
Dr Marks’ Fig 9 set out the absolute rate of myocardial infarctions for patients taking naproxen, as drawn from various studies, including VIGOR. It did not set out the rate relative to patients taking placebo (ie, nothing). However that may be, it is apparent from the extract set out above that Dr Marks took no issue with Juni on this aspect. As I read his witness statement, Dr Marks’ commentary on the McGettigan article was confined to the following:
McGettigan reports results in her Table 3 from 50 computed [odds ratios] from 13 different retrospective studies, 31 analyses for VIOXX and 19 analyses for other COX-2 drugs. Based on the criterion established earlier for accepting [odds ratios] as relevant, two of the reported analyses (4%) met the criterion. This small number of statistically significant analyses would be expected to occur simply by chance when adjusting for multiplicity, so the McGettigan report provides no evidence of a true enhanced VIOXX CV risk.
Again, I do not understand this passage as addressing McGettigan to the extent that Prof Woodward relied on it for his naproxen analysis. As it happens, in his responsive witness statement Prof Woodward agreed with Dr Marks that the Juni and McGettigan studies were “not adequate”. It was for that reason, he explained, that he undertook his own literature search and meta‑analysis.
365 Dr Marks’ direct criticism of Prof Woodward was contained in the following paragraph:
The epidemiological studies are mixed regarding naproxen protection, but the RCT results shown in my Figure 9 most clearly document that naproxen results have been quite similar to the results expected for aspirin usage. I would note that Professor Woodward would have had access to the White (2003) and Farkouh (2004) publications to evaluate published naproxen RCT information.
I note that there was no criticism of the methodology used by, nor of the technical reliability of the results reported by, Prof Woodward on this issue. In his responsive witness statement, Prof Woodward interpreted Dr Marks’ observation that the epidemiological studies were mixed as indicating that some showed a cardioprotective effect for naproxen, while others did not. Prof Woodward agreed, adhering to his conclusion that, by his cumulative pooled analysis, a small cardioprotective effect was apparent. He added that he did not include Farkouh (2004) because it did not have a placebo arm and he did not include White (2003) because the results comparing naproxen to placebo had never been published. Prof Woodward was not cross‑examined on the adequacy of these responses.
366 There are some studies on Prof Woodward’s list which appear to demonstrate the cardioprotective effect of naproxen (as against placebo) at a statistically significant level – Watson, Solomon and Kimmel. Prof Woodward confirmed that the relative risk (point estimate) figure of the Watson study represented a 39% reduction in risk from taking naproxen rather than placebo. He confirmed also that the 95% confidence limits for the risk reduction were 6% and 61%. That is to say, one could be 95% confident that the true reduction in risk fell between 6% and 61%. Prof Woodward agreed with counsel for the respondents that, standing alone, the Watson study would “shine a light that said naproxen is cardioprotective”. Likewise with the Kimmel study: the point estimate for the risk reduction was 52% within 95% confidence limits of 18% and 72%. The present relevance of these studies (and of the Solomon study) is that they demonstrate, in a statistically valid way, an association between the consumption of naproxen and a reduced incidence of myocardial infarction and that, at least in some patient groups and under some circumstances, naproxen has the potential to be cardioprotective to a considerable degree. Prof Woodward’s analysis may well show that, over a number of observational studies, the cardioprotective effect of naproxen was the order of 2% only, but that conclusion does not, in my opinion, exclude the prospect that the beneficial effect of naproxen was responsible for a large measure of the excess of myocardial infarctions observed in the Vioxx arm of VIGOR. It is, in the circumstances, necessary to turn to the cardiological evidence.
367 In their joint report, the cardiologists noted that there were no randomised placebo‑controlled trials studying the risk of thrombosis with naproxen compared to placebo. They added that some observational studies were consistent with the existence of “a small benefit”, while others suggested that there was no benefit. However, they expressed the view that naproxen had “a significant anti‑platelet effect at a dose of 500 mg twice daily … similar to that of low-dose aspirin”. They referred to Capone (2004). In that article (which reported on a pharmacological study), the authors stated:
We found that the chronic administration of a therapeutic anti‑inflammatory dose of naproxen (500 mg BID) to healthy subjects caused persistent and almost complete suppression of platelet TXB2 production throughout the 12‑hour dosing interval that was indistinguishable from that of low‑dose aspirin (100 mg/d). However, whereas aspirin pharmacokinetics is dissociated from pharmacodynamics and 100 mg represents approximately a 3‑fold excess versus the lowest effective dose to saturate platelet COX-1 activity, naproxen pharmacodynamics is strictly related to its systemic bioavailability, as shown by the significant recovery of platelet COX-1 activity at 24 hours after dosing. Moreover, 500 mg BID is probably close to but not quite at the top of the dose‑response curve for COX-1 inhibition. This suggests that compliance and daily dose are likely to represent the main determinants of the clinical efficacy of naproxen for cardiovascular protection. Thus, the apparently conflicting results of a randomized clinical trial, like VIGOR, and observational studies may reflect markedly different rates of compliance and regular use of a high dose of the drug in the 2 settings.
The “observational studies” mentioned in the final sentence of this extract were footnoted as those of Mamdani, Solomon, Watson, Rahme and Ray (Jan 02), all of which are on Prof Woodward’s list set out in para 351 above. In the cardiologists’ joint report, Profs Celermajer and Vaughan said that the dose and frequency of taking naproxen may be important in evaluating the observational study data. In his evidence in concurrent session, Prof Celermajer referred to the observational studies that had suggested that naproxen had a protective effect of the order of 10% or less, and continued:
I believe the most likely explanation for that is that people cannot take naproxen regularly other than when they are in a clinical trial because it’s a very difficult medication to take regularly because of tummy upset, and I think the most likely scenario is that in observational studies people only take it when they have pain and they don’t derive the antiplatelet benefit because to do that you have to take it twice daily, as was the supervised and complied with case in the VIGOR study.
He subsequently made what he described as “a really important point”:
[A]ll arthritis drugs work by inhibiting COX-2. They are all given in a dose to inhibit COX-2 because that’s what causes the pain of arthritis. That’s why Celebrex is given at 200 milligrams and Vioxx at 25 milligrams, because it doesn’t matter where on the IC50 curve they sit, you give enough to knock COX-2. So they are all acting as COX-2 inhibitors. It’s a question of how much they spare COX-1. So they are better regarded as COX-1 sparers rather than COX-2 inhibitors. So the IC50 is balanced out by the dose at which they are given which means that the traditional NSAIDs vary by their COX-1 sparing.
In support of this explanation, Prof Celermajer relied on the Capone article, as well as a diagram reproduced in Weir et al which was effectively the same as the one I have set out at para 91 above. As I understand it, the data from which that diagram was prepared were those of the Merck Protocol 061.
368 What Prof Celermajer described, “the IC50 curve” was a reference not to a curve as such (as I understand it) but to a statistic which may be assigned to any agent which inhibits COX-1, COX-2 or both. It is the number yielded when the concentration of the agent in the blood required to inhibit the activity of COX-1 by 50% is the numerator and the concentration of the agent in the blood required to inhibit the activity of COX-2 by 50% is the demonator. The number for various NSAIDs are shown in Baigent (2003) as follows:
|
Inhibitor |
COX-1:COX-2 IC50 ratio |
|
Ibuprofen |
0.5 |
|
Naproxen |
0.7 |
|
6-MNA (active metabolite of nabumetone) |
1.5 |
|
Acetaminophen |
1.6 |
|
Indomethacin |
1.9 |
|
Meloxicam |
18.0 |
|
Nimesulide |
19.0 |
|
Diclofenac |
29.0 |
|
Celecoxib |
30.0 |
|
Rofecoxib |
267.0 |
A low value will be yielded by an agent which is highly selective for (ie to inhibit) COX-1, and a high value will be yielded by an agent which is highly selective for COX-2.
369 Profs Zipes and Harper relied upon the studies examined by Prof Woodward, and upon his analysis, to conclude that the cardiovascular protective effect of naproxen was in the order of 10% or less. They made a distinction between the antiplatelet effect, on the one hand, and the cardioprotective effect, on the other hand, of naproxen. Prof Harper said:
It may well be true that naproxen has a similar antiplatelet activity to aspirin but that doesn’t mean it necessarily has a similar degree of protection against thrombosis because there may be other factors, for example on how much prostacyclin is around in the setting of two drugs. So even though there is a similar antiplatelet action, the human data suggesting only a less than 10 per cent protective effect suggests that for one reason or other the antithrombotic effect is not as powerful as aspirin.
It is on this basis that Prof Harper, and, as I understood him, Prof Zipes also, justified their concurrence in the view that naproxen taken twice daily in a dose of 500 mg had an antiplatelet effect similar to that of low‑dose aspirin, with their conclusion that cardiovascular benefit to be had from the consumption of naproxen was at best 10%. In this respect they had, it seems to me, some support from the following reservation expressed in the conclusion of the Capone (2004) article:
[T]he clinical relevance of simultaneous suppression of TXA2 and PGI2 by naproxen in patients with cardiovascular disease remains to be determined. Moreover, the impact of naproxen on COX-2‑dependent sources of thromboxane biosynthesis might contribute to its clinical effects in preventing myocardial infarction.
370 I do not, however, consider that making a distinction between the antiplatelet and the cardioprotective effects of naproxen is entirely an answer to Prof Celermajer’s point. That some of the observational studies revealed a significant (and, in the cases of Watson and Kimmel, quite strong) diminution in the occurrence of myocardial infarctions amongst those taking naproxen compared to placebo is evidence of the cardioprotective effect of naproxen in vivo (ie not merely of the antiplatelet effect). It is when one asks why this would have been so, and why there is so much variation as between the results of the observational studies on Prof Woodward’s list, that one turns to the pharmacological studies (such as Protocol 061 and Capone(2004)). Then one discovers that non‑selective NSAIDs do have an antiplatelet action to a degree (as they must, by inhibiting COX-1) and that the duration of that action varies considerably as between NSAIDs. As Dr Reicin explained in her evidence, the action of non-aspirin NSAIDs upon COX-1 is reversible, and thus depends on their concentration in the blood. The pharmacological studies tell us that naproxen has an effect on platelets over about 12 hours: it seems to be the longest‑lasting NSAID. It is here that the dosing interval becomes critical: the ingestion of naproxen twice daily will, it seems, maintain the concentration of the drug in the blood at such levels as will inhibit COX-1 to the same, or to about the same, extent as aspirin. I accept Prof Celermajer’s evidence that this circumstance most likely explains the widely differing results of the observational studies.
371 It follows that the respondents have established that, if taken at a dose of 500 mg twice daily (the dose which was used in the VIGOR trial), and if so taken conscientiously, naproxen is protective, to a degree, against myocardial infarction. However, is that degree sufficient to explain the disparity of myocardial infarctions (to take the applicant’s strongest case) as between the two arms in VIGOR? Such a question was answered categorically in the negative by experts called on behalf of the applicant. Prof Zipes said that a protective effect of naproxen, even if the equivalent of that of aspirin, could not account for a relative risk of 5. Prof Madigan said that he was unaware of any scientific study in which a cardioprotective effect of naproxen sufficiently large to explain the VIGOR results had been claimed. Having concluded that naproxen produced about a 2% reduction in the risk of myocardial infarction, Prof Woodward said:
At no time could naproxen be reasonably assumed to have anything like the 80% risk reduction that was postulated in VIGOR, and it was unreasonable for Bombardier et al (2000) to suggest that the excess cases of [myocardial infarction] in the rofecoxib group was purely due to cardiovascular protection conferred by naproxen.
In the light of the findings which I have made above, I would not limit the potential cardioprotective effect of naproxen to 2%, but the significance of the evidence of Prof Woodward and the other experts lies in the response which that evidence drew from Dr Reicin.
372 In her witness statement in reply, Dr Reicin dealt with the question whether, assuming naproxen to have a potentially cardioprotective effect, that effect was large enough to explain the VIGOR results. She said that this was an issue that MRL considered when it interpreted those results. She said that because Dr FitzGerald hypothesized that Vioxx may be pro‑thrombotic – in effect an anti‑aspirin – it was logical to look at aspirin studies by way of comparison with the effect of naproxen as seen in VIGOR. In relation to the APTC combined endpoint, Dr Reicin said that it was known that consumption of aspirin was associated with a risk reduction of 25% (referring in this regard to APTC (1994)). She then noted that the 95% confidence interval for the reduction in the risk of suffering an APTC event from taking naproxen rather than Vioxx in the VIGOR trial was 9% – 71%. Dr Reicin noted that 25% fell within this range, concluding that “statistically the reduction observed with naproxen in VIGOR is not inconsistent with what has been observed with aspirin”.
373 Dr Reicin was cross‑examined extensively on this line of reasoning. Senior counsel for the applicant paid little attention to the APTC endpoint, but went directly to the focus of the comments made by Profs Zipes and Woodward, namely, the size of the myocardial infarction risk differential observed in VIGOR. When the 1994 study was examined in the course of cross‑examination, it emerged that the risk reduction (from taking aspirin) in the case of myocardial infarction in that study was of the order of 34%. It was pointed out on behalf of the applicant that this figure did not fall within the 95% confidence interval for the reduction in the risk of myocardial infarction from taking naproxen rather than Vioxx as found in VIGOR, which was 42% – 93%. Further, counsel for the applicant submitted that, on the basis that the 95% confidence interval was ± twice the standard deviation, and the standard deviation in the APTC data was 3%, the lower end of the 95% interval of the risk reduction in VIGOR (42%) was still greater than the upper end of the 95% of the risk reduction achieved by aspirin (34% + 6% = 40%). Neither Dr Reicin in her evidence, nor the respondents in their submissions, came fully to grips with the position articulated by counsel for the applicant in these respects.
374 The analysis upon which Dr Reicin relied to demonstrate that the aspirin risk reduction for the APTC endpoint lay within the 95% confidence interval for the corresponding naproxen risk reduction in VIGOR thus shows that the aspirin risk reduction for myocardial infarction lies outside the 95% confidence interval for the corresponding naproxen risk reduction in VIGOR. That is to say, we may be 95% confident that the reduction in risk of myocardial infarction arising from taking naproxen rather than Vioxx is greater than 42%, whereas the figure accepted by Dr Reicin as representing the risk reduction arising from the consumption of aspirin is 34% and, with 95% confidence, no more than 40%. Clearly the 1994 study provides no support for the proposition that, in relation to myocardial infarction, the whole of the difference observed as between the two arms of the VIGOR trial is to be explained by the cardioprotective effect of naproxen.
375 Neither does the other evidence upon which the respondents relied provide any support for a conclusion that the cardioprotective effect of naproxen was sufficient to explain the 5‑fold difference in the occurrence of myocardial infarctions observed in VIGOR. In their written submissions, the respondents relied upon the latter part of the following extract from Baigent (2003) (I have included some text immediately preceding the passage referred to by the respondents to provide context):
Since naproxen produces less consistent inhibition of TXA2 than does low‑dose aspirin, the degree of protection against myocardial infarction achieved by naproxen is unlikely to be greater than that produced by low‑dose aspirin, but could be broadly comparable in size. The most recent report from the Antithrombotic Treatment Trialists’ (ATT) collaboration showed that antiplatelet therapy reduced the risk of myocardial infarction by approximately one‑third among high‑risk patients [ATT (2002)], and the reduction observed in a low‑risk setting, reported in a recent meta‑analysis of trials comparing aspirin and placebo, was similar (30% reduction in risk, 95% CI 21–38%) [Sanmuganathan (2001)]. Thus, naproxen could reduce myocardial infarction by up to one‑third, as compared with the effects of rofecoxib. In the VIGOR trial, naproxen, as compared with rofecoxib, was associated with an implausibly large 80% (95% CI 42–93%) lower risk of myocardial infarction; if naproxen does have a substantial antiplatelet effect, then the lower end of this confidence interval is not too dissimilar to the upper end of the confidence interval for the effects of aspirin in a low‑risk setting.
As I understand the burden of the evidence of the biostatisticians in the present case, the relationship which the authors here described as “not too dissimilar” might more accurately be said to exclude, with 95% confidence, the possibility that the naproxen effect seen in VIGOR was no larger than the aspirin effect seen in the studies to which the authors referred.
376 Prof Celermajer did not suggest that the 5‑fold differential in myocardial infarctions observed in VIGOR might wholly be explained by the cardioprotective work of naproxen. As I have noted above (para 348), his objection to Prof Woodward’s analysis was that it focussed on one outcome only. However, it is the risk of suffering any one of the pleaded cardiovascular conditions that must concern the court in the present case. The 5‑fold discrepancy to which Prof Woodward referred must be addressed, and cannot be sidestepped on the basis proposed by Prof Celermajer. Prof Vaughan also gave evidence on the subject of the extent of cardioprotection presumptively available from naproxen, but that related specifically to individuals (such as the patients in VIGOR) with a “systemically inflammatory state”. That evidence is more directly relevant to Dr Reicin’s next point, to which I shall turn. Absent such considerations, however, the position appears to be that low‑dose aspirin, by its antiplatelet effect, has the potential to reduce the incidence of myocardial infarctions by up to about a third, and that naproxen, regularly taken twice daily, is capable of achieving a similar result. Manifestly, this conclusion does not explain the full extent of the disparity between the two arms in the VIGOR trial.
377 The second explanation for that disparity offered by Dr Reicin in her statement in reply was the following:
[T]he magnitude of effect of naproxen observed in VIGOR may also have been in part a function of the rheumatoid arthritis population that was studied. Rheumatoid arthritis is a chronic inflammatory disease associated with elevated levels of C‑reactive protein, a marker of systemic inflammation. As explained in the 2005 FDA Advisory Committee Background Information package prepared by Merck:
Studies have suggested that aspirin has a larger relative benefit in higher risk patients defined either by levels of C‑reactive protein (CRP) or as defined clinically. In the Physician’s [sic] Health Study, the risk reduction for myocardial infarction ranged from 13.9% in the quartile of patients with the lowest level of CRP to 55.7% in the quartile with the highest CRP levels. In the antiplatelet drugs meta‑analysis, a greater risk reduction was seen in higher risk patients (37% reduction of the APTC combined endpoint in patients with coronary artery disease and ~50% reduction in patients with unstable angina or post‑angioplasty). The patients in VIGOR all had [rheumatoid arthritis], [rheumatoid arthritis] patients generally have higher CRP levels than patients without inflammatory disease, and [rheumatoid arthritis] patients have an increased risk of coronary artery disease and are a recognized high risk group for coronary artery disease.
The study to which Dr Reicin referred was published as Ridker (1997). There the researchers grouped the patients being studied into quartiles, according to their C‑reactive protein values. They found:
The use of aspirin was associated with significant reductions in the risk of myocardial infarction (55.7 percent reduction, P = 0.02) among men in the highest quartile but with only small, nonsignificant reductions among those in the lowest quartile (13.9 percent, P = 0.77).
378 Here it is convenient to mention a circumstance that appeared to be fairly uncontroversial, namely, that rheumatoid arthritis sufferers as a group are especially susceptibe to CVT disease. This was the subject of comment by the rheumatologists in their joint report. They noted that multiple studies had shown an increased risk for serious CVT events in patients with rheumatoid arthritis. Dr Bertouch said:
If you have rheumatoid arthritis your risk of ischaemic heart disease is doubled from the time that that diagnosis is made. If you have rheumatoid arthritis for more than 10 years the figures tell us that your risk of coronary artery disease is tripled ….
Counsel for the respondents put to the rheumatologists the proposition that, in this susceptible group, the cardioprotective effect of naproxen might be even greater than amongst the population generally. Prof Cleland did not accept it. He said:
Well, not necessarily. I mean, we don’t have a strong basis for arguing there is a cardioprotective effect of naproxen at all. So that to the extent that any agent might have a real cardioprotective effect, the general argument is that you are more likely to see that effect if their mechanism of action is relevant in a population with a high risk rate – with a high event rate. It’s just a numbers business of getting enough events to get statistical significance.
The point being made in the latter section of this evidence was that the greater number of events in a trial involving rheumatoid arthritis patients would lead to the more ready achievement of statistical significance. Dr Bertouch answered counsel’s question with the words, “logically that would follow”.
379 The issue, of course, is not whether rheumatoid arthritis sufferers are especially susceptible to CVT disease: patients in both arms of the VIGOR trial were rheumatoid arthritis sufferers. The issue is whether such persons are likely to derive unusually high levels of cardioprotection from antiplatelet agents such as aspirin or, presumptively, naproxen. Prof Vaughan thought that a distinct possibility. Of the 1997 physicians’ health study (Ridker 1997), and its relevance in the context of the VIGOR results, he said:
[O]ne of the most important pivotal trials that helped us incorporate aspirin into our everyday therapeutic lives was the physicians health study. It turns out aspirin really only – the preponderance of the effect of aspirin was seen in a group of patients, group of individuals that had evidence of systemic inflammation, had elevated levels of the high sensitivity c‑reactive protein. Other groups of physicians that did not have systemic inflammation had much less effect or no effect. So it’s not surprising to me that the group in VIGOR, a group with rheumatoid disease, a group with a systemic inflammatory state responded quite nicely to a potent anti‑inflammatory drug with potent platelet effects because they are a group that responds very well. So I think we are seeing an effect that’s greater than 10 per cent in that group. I don’t know exactly what the benefit might have been. I think it could be as high as a 50 per cent reduction. It could be 20, it could be 30. I don’t know exactly what it is.
So far as I can see, this was the first time that Ridker (1997) was referred to in the evidence. As mentioned, it was referred to by Dr Reicin only in her statement in reply. However, as I have noted above (para 129), the possibility was canvassed in the Clinical Study Report for VIGOR (although Ridker (1997) was not referred to).
380 In this part of my reasons I am concerned with the objective likelihood of the VIGOR myocardial infarction results being wholly explicable by the cardioprotective effect of naproxen. In the evidence to which I have referred, neither of the respondents’ cardiologists nor, for that matter, Dr Reicin herself, was prepared to affirm the proposition that some part of the excess of myocardial infarctions in the VIGOR trial was due not to the beneficial effect of naproxen as such but an exaggerated effect arising from the particular susceptibility of rheumatoid arthritis suffered to CVT disease. I am not disposed to take Prof Vaughan as going further than to suggest that, in patients with systemic inflammation, the cardioprotective effect of naproxen may be more than the one‑third proposed by APTC (1994) and Baigent (2003), and may possibly be up to about one‑half. I do not understand Prof Vaughan to have offered his professional opinion that it was, however. Dr Reicin went no further than to opine that the effect of naproxen observed in VIGOR “may also have been in part a function of the rheumatoid arthritis population that was studied”. Prof Celermajer pointed out that patients with rheumatoid arthritis “have a systemic pro‑inflammatory state which is documented to place them at unusually high risk of [cardiovascular] events”, but did not deal with the possibility that the extent of cardioprotection provided by aspirin, and therefore (presumptively) by naproxen, was greater amongst such patients than in people generally.
381 In the circumstances, I am not prepared to find, favourably to the respondents, that it was not only possible but likely that part of the observed discrepancy in the myocardial infarction results in the VIGOR trial was due to the especially pronounced beneficial work of naproxen in the context of patients with a systemically inflammatory condition. I stress that the point here is not whether such patients are especially susceptible to cardiovascular disease. If they are, it would not be surprising to see elevated event rates in both arms of a trial such as VIGOR. Rather, the point is whether the size of the differential in event rates as between the two arms in VIGOR should be explained in part by the circumstances that all patients, as rheumatoid arthritis sufferers, were presumably in such a condition. The evidence does not persuade me that it should.
382 The third explanation offered by Dr Reicin in her statement in reply related to Prof Woodward’s analysis of the observational studies (see paras 351‑363 above) on which he had relied to conclude that the cardioprotective effect of naproxen was about 2%. Dr Reicin pointed out that it should not be assumed that the patients whose clinical experiences were observed in such studies were compliant in taking naproxen to the same extent as were, she asserted, those who participated in the VIGOR trial. I do not make that assumption. Indeed, I have effectively accepted the proposition that VIGOR might well, because of the rate of compliance adhered to by the patients in the naproxen arm, be an example of a trial in which near complete (ie in the range 90‑95%) inhibition of platelet COX-1 was achieved. But the result of accepting that is to allow for the realistic prospect that naproxen might, with proper compliance, be about as cardioprotective as aspirin. It does not take one the distance required to explain the VIGOR myocardial infarction results.
383 For reasons which I have attempted to explain, I am not satisfied that the cardioprotective effect of naproxen was sufficient to explain the myocardial infarction data yielded by the VIGOR trial. This conclusion is not inconsistent with the view expressed by Profs Celermajer and Vaughan in the joint cardiology report. They were of the view that the naproxen hypothesis was a “biologically plausible explanation” for the VIGOR findings. They added:
Many authors … have written that a naproxen effect and the play of chance can explain the VIGOR findings, without needing to invoke an adverse cardiovascular effect of Vioxx. … We believe that this was then and remains now a reasonable interpretation as a contributing factor in the VIGOR results.
I read this as relying upon, first, the observational studies that had shown that naproxen was associated with a substantial reduction in the incidence of myocardial infarctions, and secondly, the pharmacological studies that had shown that, under certain conditions of dosage and compliance, naproxen appeared to have an antiplatelet effect the equivalent of that of low‑dose aspirin, but as recognising that even those circumstances taken together could not be treated as more than a contributing factor in the VIGOR myocardial infarction results. Those circumstances did not go the full distance to explain those results.
384 Save for those with which I have already dealt, there were two other circumstances mentioned in the evidence that might have contributed to the VIGOR results. The first was “the play of chance”. This circumstance had two dimensions. The first was the prospect that the play of chance was responsible for a statistically significant excess of the occurrence of the events of interest in the Vioxx arm compared to the naproxen arm. Ultimately the respondents did not submit that the results might have been explained by the play of chance in this sense. Dr Reicin did not think that they were. Indeed, as Prof Madigan pointed out in a different context, we know in this kind of situation what the play of chance was: it was 5 chances in 100. As a court exercising civil jurisdiction, I should not accept the play of chance as a probable explanation for a significant excess, say, of myocardial infarctions in the Vioxx arm of the VIGOR trial.
385 The other dimension of the problem is what Prof Celermajer described as “the fragility of the best estimate”. He said:
We are talking about the fragility of the best estimate, so the best estimate of 20 versus 4 is 5.0 but how confident can you be that 5.0 is the real number so if I may wrap some numbers around it. Say, for example, if two of the events in the Vioxx group had happened in the naproxen group instead of 20 to 4 it would be 18 to 6 and that would make the 5 into a 3. That’s not what happened, it was 20 to 4 and it was 5.0 but when you are dealing with very low numbers, very subtle shifts have major effects on the best point estimate. So the play of chance referred to by Patrono certainly can have an effect when you do the study only one time.
The reference to which Prof Celermajer referred as “Patrono” was Patrono (2004), a paper prepared for the Seventh American College of Chest Physicians Conference on Antithrombotic and Thrombolytic Therapy: Evidence‑Based Guidelines. The authors of that paper (who included Dr FitzGerald), under the heading “Coxibs and cardiovascular disease”, said:
While the cause of the apparent excess risk of [myocardial infarctions] in the [VIGOR] trial cannot be conclusively established, a combination of some cardioprotective effect of naproxen and the play of chance does seem to offer a plausible explanation for these unexpected findings. While other mechanisms cannot be discounted, there is currently little evidence in humans to support a prothrombotic effect for coxibs.
Prof Celermajer’s observations are, as I read them, related to the circumstance, explained by the biostatisticians, that we can be 95% confident that the true relative risk of suffering a myocardial infarction from taking Vioxx rather than naproxen, for a population with the characteristics of the patients in the VIGOR trial, lay between 1.72 and 14.29. Had the point estimate been 3.00 rather than 5.00, the whole confidence interval may still have been situated above unity, but then the potential for the cardioprotective effect of naproxen to have explained substantially all of the difference might realistically have come into play.
386 While the circumstance to which Prof Celermajer referred is clearly relevant to an understanding of the clinical ramifications of the VIGOR results, concerned as I am at present with the question whether those results were consistent with APPROVe, I do not think I am in any position to speculate as to how things might have been if they were otherwise. I recognise that the numbers were small, but (mercifully) that seems nigh inevitable when one is concerned with serious events such as myocardial infarctions. I do not think I should allow scope for consideration of the play of chance in this second sense referred to by Prof Celermajer (although the point is not thereby rendered irrelevant to the considerations which Merck might properly have taken into account in understanding the VIGOR results – a matter to which I shall come later in these reasons).
387 The other circumstance was dose. In VIGOR, the dose of Vioxx taken by patients was 50 mg daily. This was twice the dose taken in APPROVe. The influence of dose upon the observed relationship between the ingestion of Vioxx and the occurrence of CVT events is the subject of another of the Bradford Hill criteria, and I return to it below. It may be noted at this point, however, that, in the cardiology concurrent session, Profs Zipes, Celermajer and Vaughan said that VIGOR told us nothing about a situation in which someone was taking Vioxx at 25 mg daily. Prof Harper said that VIGOR told us that, at a dose of 50 mg, Vioxx probably has cardiac toxicity, adding: “It doesn’t necessarily mean it would at a lower dose but it doesn’t exclude that possibility, obviously, either.” Because of the potential significance of dose to an understanding of the VIGOR results, I propose to leave any conclusion on the consistency of VIGOR with APPROVe until after I have dealt with other aspects of the applicant’s case on risk.
The Merck arthritis studies
388 Still on the subject of the consistency of the APPROVe results with those of other trials and studies, the applicant next relied upon the results of 21 placebo‑controlled clinical trials (of four weeks’ duration or longer) involving patients with arthritis conducted by Merck between 1995 and 2003. They included the osteoarthritis trials referred to in para 87 above and the arthritis trials included in the pooled analysis referred to in paras 143ff above. However, the applicant did not accept Merck’s treatment of the arthritis data. His case in this regard was based upon a complete re‑analysis undertaken by Prof Madigan. The validity of that re‑analysis was put in issue by the respondents, who supported the approach taken originally by Merck and published in Konstam (2001).
389 Prof Madigan introduced his expert’s report in the following terms:
The VIGOR results became available in March 2000 and suggested that Vioxx posed a significant cardiovascular risk as compared with an active comparator, naproxen. My analysis below suggests that data from placebo‑controlled trials conducted before, during, and after VIGOR strongly support the proposition that those results largely reflect the cardiovascular thrombotic toxicity of Vioxx rather than naproxen cardioprotection and establish to a reasonable degree of scientific certainty that Vioxx causes an array of cardiovascular adverse events.
390 Prof Madigan considered first the arthritis trials of Vioxx. He said that, between 1995 and 2003, Merck conducted 21 placebo‑controlled clinical trials of four weeks’ duration or longer studying Vioxx in patients with arthritis. In his view, these trials were of particular importance, first because they were placebo‑controlled, secondly because Vioxx was most commonly used to treat patients with arthritis, thirdly because the relative homogeneity of the patient populations provided a justification for within‑block pooling (in which respect he referred to Merck’s own analysis strategy of January 1999 which, according to him, specifically called for a combined analysis of osteoarthritis and rheumatoid arthritis placebo‑controlled studies separate from the Alzheimer’s studies), and fourthly because the early commencement date of these trials (1995) enabled him to characterize the available evidence concerning CVT toxicity at crucial stages in the development of Vioxx.
391 Prof Madigan based his analysis upon the “Rofecoxib Cardiovascular Combined‑Analysis Update” prepared by Merck in 2003, the version of which in evidence is dated August of that year. Table 13 in that document compared Vioxx to placebo by reference to CVT events, and was the same as the like table included in the report sent to the FDA in Merck’s fourth pooled analysis on 22 March 2004: see para 187 above. It will be recalled that the data in that table did not reveal, at a statistically significant level, that there was a relative risk of encountering a CVT event by taking Vioxx instead of placebo. Prof Madigan added to the data and modified the analysis there presented in the following ways. First, he took account of the results of six studies which had not been included in Table 13. Of these, Prof Madigan did not know why Protocols 010 and 017 had been omitted by Merck. The remaining four – Protocols 112, 116, 219 and 220 – had apparently been omitted because they did not provide comparisons of Vioxx to non-selective NSAIDs. Secondly, he added the osteoarthritis and rheumatoid arthritis data together to make a single block for which he calculated the relative risk. In Table 13, these data were not aggregated in this form: the only relative risk figures reported related to the osteoarthritis and rheumatoid arthritis comparisons separately, and to the whole body of data, including those arising from the Alzheimer’s disease studies.
392 The relative risk of encountering a CVT endpoint among arthritis patients, from taking Vioxx rather than placebo, as calculated by Prof Madigan was 2.8 (1.2, 7.4). He undertook a like calculation, but this time including only the studies listed in Table 13 (ie excluding the six additional studies not reported on by Merck). There the risk was 3.1 (1.3, 9.2). In each case p = 0.01.
393 Prof Madigan next examined the combined osteoarthritis and rheumatoid arthritis data as they stood as at 31 December each year from 1997 to 2003. Here he used all the studies that were available to Merck at each point, including the six which were not part of Table 13. His table of these data was the following:
|
|
Vioxx |
Comparator |
|
|
||
|
End date for Study inclusion |
N |
PYR |
N |
PYR |
Relative risk & 95%CI |
p‑value |
|
31 Dec 1997
|
9 |
207 |
1 |
50 |
2.4 (0.4, 56.0) |
0.4 |
|
31 Dec 1998 |
17 |
539 |
4 |
169 |
1.4 (0.5, 4.8) |
0.6 |
|
31 Dec 1999 |
25 |
706 |
4 |
248 |
2.2 (0.8, 7.5) |
0.1 |
|
31 Dec 2000 |
31 |
1173 |
5 |
475 |
2.4 (1.0, 6.9) |
0.06 |
|
31 Dec 2001 |
no change |
no change |
|
|
||
|
31 Dec 2002 |
32 |
1268 |
5 |
676 |
3.4 (1.4, 9.8) |
0.005 |
|
31 Dec 2003 |
32 |
1304 |
6 |
692 |
2.8 (1.2, 7.4) |
0.01 |
Prof Madigan made the point that, had Merck undertaken this analysis in December 2000 – which it well might have – it would have come up with a risk of CVT events which was marginally significant statistically and which could only have been interpreted as “an ominous trend”, particularly when taken with the results of VIGOR. By the end of 2002, the risk was apparent at a statistically significant level, and it remained that way a year later. Prof Madigan also undertook alternative analyses which, he claimed, Merck might have undertaken itself, but he expressed the view that the results of them were “qualitatively the same” as those set out above.
394 “For the record”, as he put it, Prof Madigan also provided the results which would have been given in place of those in Table 13 if all investigator reported events (ie not only those which were confirmed on adjudication) had been included and the data from all 21 studies were used. He came up with relative risk figures for the osteoarthritis studies of 1.82 (0.78, 5.00), for the rheumatoid arthritis studies of 3.23 (0.53, 83.4), and for the combined osteoarthritis and rheumatoid arthritis studies of 2.10 (0.95, 5.18). None of those risk figures was significant, but Prof Madigan expressed the view that “the evidence points in a disturbing direction”.
395 Dr Reicin justified Merck’s exclusion of the six studies from its pooled analysis. She said that Protocols 010 and 017 were conducted at a time when Merck had not yet determined the appropriate therapeutic dose for Vioxx. They included daily doses (in the case of Protocol 010) of 25 mg and 125 mg and (in the case of Protocol 017) of 125 mg and 175 mg. The object of Merck’s pooled analyses was to study the effect of Vioxx taken at therapeutic doses. Dr Reicin explained that the other four protocols that were omitted involved comparisons of Vioxx with celecoxib, not with non‑selective NSAIDs. However, it seems that these studies did include placebo arms, and I must say that no entirely satisfactory explanation of why the placebo/Vioxx data might not have been included was provided by the respondents. Notwithstanding these reservations, the applicant was content to rely on so much of Prof Madigan’s analysis as did not include these six studies, pointing out that it yielded a statistically significant risk of taking Vioxx which was actually greater than the figure which resulted from the exclusion of them.
396 Dr Reicin also justified Merck’s use of an overall risk figure – one which was based on data from the Alzheimer’s trials as well as from the arthritis trials – in its pooled analyses. She said that to have looked at the placebo‑controlled data from the osteoarthritis and rheumatoid studies in isolation would have been less informative than what Merck actually did, which was to consider “the totality of the data”. Dr Reicin was unaware of any biological or medical reason that made it inappropriate to include the placebo data from the Alzheimer’s trials when conducting the pooled analysis. From a biological standpoint, if Vioxx increased the risk of CVT events among patients with arthritis, she would have expected to see that risk in the Alzheimer’s trials as well. Neither, in her view, was there any statistical reason which made it inappropriate to pool placebo data from the Alzheimer’s, osteoarthritis and rheumatoid arthritis trials. The data were tested for heterogeneity (a test used to determine whether or not it is statistically appropriate to pool data from different sets), and no significant heterogeneity was seen across indications. Thus Dr Reicin thought that the data were sufficiently similar for valid pooling. There was no serious challenge to this evidence: it was not, for example, suggested that there was a medical or biological reason why the data should not be pooled, or that a test for heterogeneity would not have produced the results referred to by Dr Reicin.
397 The respondents submitted that Prof Madigan’s analysis of the osteoarthritis and rheumatoid arthritis studies “was based on such low absolute numbers of CV events in the OA and RA placebo arms that any statistical comparison to Vioxx will be imprecise and must be interpreted with caution”. This submission was supported only by a reference to evidence (subsequently) given by Prof Celermajer with respect to a different study. It was not put to Prof Madigan that his comparison was “imprecise and must be interpreted with caution”. Neither did Dr Marks express any such reservation in his evidence. In his report, which was filed in reply to the reports of the biostatisticians called by the applicant, the only comment made which might be thought to touch the present issue was that pooling decisions should be made by medical experts without regard for the statistical results, that pooling should be based on medical relevance, and that there was no statistical reason for Merck not to have pooled the arthritis data with that from the Alzheimer’s disease trials.
398 In the result, I find it to have been established by the evidence of Prof Madigan that, amongst arthritis patients as a group, the consumption of Vioxx gave rise to a risk of encountering a CVT event which, at the 95% confidence interval, lay within the band 1.3‑9.2, with a point estimate of 3.1. In using these figures, I have taken the version of Prof Madigan’s analysis which follows Merck in excluding Protocols 010, 017, 112, 116, 219 and 220.
399 I cannot so readily accept Prof Madigan’s table of annual relative risk rates set out in para 393 above. That table was based on the data from all 21 osteoarthritis/rheumatoid arthritis studies, and I am disposed to accept Dr Reicin’s justification for the exclusion of Protocols 010 and 017. As indicated above, I have reservations about the stated justification for the exclusion of Protocols 112, 116, 219 and 220, but, absent some assistance from Prof Madigan or the other biostatisticians, I am in no position to speculate on what Prof Madigan’s table would have looked like had the data from these protocols been included, but those from Protocols 010 and 017 excluded. These issues were not developed in the concurrent evidence session of the biostatisticians, and Dr Marks said nothing about them in his report. Neither did the parties address them in their submissions: the applicant merely asserted that Prof Madigan’s table should be accepted, and the respondents merely asserted that Dr Reicin’s justifications for the exclusion of the six studies should be accepted.
The Merck Alzheimer’s disease studies
400 The applicant next submitted that Merck’s trials of Vioxx with patients with, or at risk of, Alzheimer’s disease, properly understood, disclosed a greater risk of CVT events amongst patients taking the drug. These were the studies referred to at para 146 above, and also included in Merck’s pooled analysis. It will be recalled that the relative risk of encountering a CVT event from using Vioxx rather than placebo, as disclosed in the Alzheimer’s trials, was reported in Merck’s pooled analysis at various times as 0.72 (0.42, 1.21) (July 2001), 0.86 (0.56, 1.33) (May 2002) and 1.03 (0.70, 1.53) (March 2004). Consistently with the CV SOP, these figures were based on Merck’s “modified ITT principle”, that is to say, an event was counted if it occurred while the patient concerned was taking the treatment to which he or she had been randomised, or within 14 days after discontinuation. Only events which had been sent for adjudication and confirmed as CVT events were counted.
401 Prof Madigan looked at the Alzheimer’s data in a different way. He used an unqualified ITT approach in the sense that he counted (or sought to count) all events that had occurred while the study in question was taking place. He justified this approach not so much because it was inherently to be preferred in matters of safety (as to which there is, as I shall show, scope for legitimate disagreement) as because the ITT approach had been “prespecified”. In his oral evidence, Prof Madigan said:
I saw that Merck has a definition of a cardiovascular thrombotic event so I used that definition. Then I looked to see what analysis Merck said they would do, so what analysis were preplanned, and in general in the world of clinical trials, … what is prespecified as golden; you cannot in general change the horse midstream. Before you do the trial you say, “Here is how I am going to analyse the data”. You conduct the trial and then you analyse the data as you said you would. So in the initial protocols, specifically for the Alzheimer’s studies, it called for an intention to treat analysis. ... You randomise someone to Vioxx, you randomise someone to placebo. You follow that patient and you see what happens to them, regardless of whether they continue to take the drug to which they were assigned. So that’s a very standard analysis, particularly for efficacy. In the context of safety, there’s more debate about the appropriateness of ITT, the notion being that it’s conservative. So it has the effect generally of diluting signals because not everyone is taking the drug that they were assigned and there is a concern, a legitimate concern, that for safety that’s not what you want to do. So there is some debate about that. But be that as it may, ITT was prespecified. So when I looked at the published analysis, particularly of the Alzheimer’s studies, I could not find an ITT analysis so it seemed to me, and it seems to me sitting here right now that that analysis needed to be done. So Merck had set up this standard operating procedure for adjudicating CVT events but there was a fundamental problem, as far as I was concerned, which was they only adjudicated events that occurred on drug or within 14 days of taking the drug, except for deaths, so that meant if a given patient was taking Vioxx, they had, let’s say, a heart attack on day 15 after they stopped taking the drug, that event was never sent for adjudication and hence, if you do an analysis of confirmed events it’s simply not counted; it’s as if it never happened. So there is a fundamental mismatch between the predefined analysis of ITT, prespecified in all three Alzheimer’s protocols, and the adjudication process.
402 A difficulty which presented itself to Prof Madigan was that Merck had sent for adjudication only those events which occurred up to the 14th day after discontinuation. He did not, therefore, have a record of confirmed CVT events collected on an ITT basis. His solution was to use investigator‑reported events, and he did so throughout, even with respect to events which had been sent for adjudication. This required him to have access to Merck’s records of investigator‑reported events and, with the help of Dr Kostis, Chairman of the Department of Medicine at the Robert Wood Johnson Medical School in New Brunswick, NJ and Dr Krumholz, Harold H. Hines Jr Professor of Medicine and Epidemiology and Public Health (Cardiology) at Yale University, and by reference to two “controlled vocabularies” used in medicine, he related those events to the items of interest in the CV SOP.
403 Prof Madigan also counted all CVT events, whereas the Merck analyses included them only if they were “serious”. He justified his approach as follows:
First, some surprising events are not tagged as serious, for example, a number of myocardial infarctions. Second, while the FDA does provide a definition of a “serious event,” some medical judgment is required to apply the definition and it is unclear that the requisite documentation to support such a judgment was always present. Third, Merck itself sometimes analyzed all “thromboembolic cardiovascular adverse event experiences” in the [osteoarthritis] trials regardless of whether the events were designated as “serious”.
404 The figure for the relative risk of taking Vioxx rather than placebo calculated by Prof Madigan in this way was 1.5 (1.1, 2.0); p = 0.02.
405 Dr Marks was most critical of Prof Madigan’s analysis of the Alzheimer’s data. In his report, Dr Marks set out in a single table a summary of Merck’s, and of Prof Madigan’s, data as follows:
Table 1. CVT results from Merck’s January 2005 FDA analysis and Professor Madigan’s original report.
------- Vioxx -------- ------- Placebo --------
n PYR PPY n PYR PPY RR (95% CI)
Merck 42 1661 2.5 48 1917 2.5 1.01 (0.67 – 1.53)
Madigan 49 2185 4.5 73 2341 3.1 1.5 (1.1 – 2.0)
PYR – Patient Years
PPY – events percentage per year (or events/100 years exposure)
406 Dr Marks then attempted to reconcile these differing data. He first identified the extent to which Merck and Prof Madigan agreed on the CVT events that had been confirmed by adjudication. This required him to add in events from Protocol 126 that were not included in the Merck report to the FDA and some events that Prof Madigan had not included but which had in fact been confirmed on adjudication. Dr Marks next looked at the events that had been included by Prof Madigan but not by Merck. These were events which had been reported as CVT events but not confirmed as such, events which occurred more than 14 days after discontinuation of drug use and events which were CVT events, but not “serious” within the meaning of the CV SOP. He expressed his conclusions by way of the following table:
Table 4. A summary of Alzheimer’s studies RRs. The first row replicates Professor Madigan’s results from his original CVT analysis. The following three rows sequentially exclude (1) events adjudicated and not confirmed as CVT events, (2) events occurring > 14 days after drug use discontinuation, and (3) non‑serious AEs. The counts also include adding the five Vioxx and seven placebo confirmed CVT SAEs not including in Professor Madigan’s original analysis. The last row updates the Merck January 2005 analysis to include confirmed CVT SAEs from protocol 126.
Vioxx Placebo
n n RR 95% CI
Madigan original analysis from Table 1 99 73 1.5 1.1 – 2.0
Madigan analysis excluding events –
Not confirmed as CVT 83 72 1.24 0.90 – 1.69
CVT confirmed > 14 days after drug use58 64 0.97 0.68 – 1.39
Non-serious AE 46 49 1.01 0.67 – 1.50
Merck 1-05 analysis adding protocol 126 48 53 1.05 0.71 – 1.54
As the heading to the table indicates, the figure of 1.05 (0.71, 1.54) is derived by adding to the analysis sent by Merck to the FDA in January 2005, which related to Protocols 078 and 091 only, corresponding data from Protocol 126. Any difference between that figure and the one sent to the FDA as part of the pooled analysis in March 2004 – 1.03 (0.70, 1.53) is not presently material.
407 It may be seen from this table that, in the Vioxx arm, 25 of the events were excluded by Dr Marks because, although they had been confirmed as CVT events, they occurred more than 14 days after discontinuation of use. There were only eight such events in the placebo arm. Dr Marks then examined these events by reference to the length of time that each patient had been taking Vioxx and the length of time after discontinuation that his or her CVT event occurred. He produced the following table:
Table 5. Summary of Alzheimer’s studies Vioxx AEs that occurred off‑drug and were counted by Professor Madigan in his analysis. Results summarize time on drug and time to event after drug discontinuation.
Follow‑up Time Off-Drug Until AE Occurs
|
Days on Drug |
1-90 days |
91-365 days |
1< years≤2 |
> 2 years |
TOTAL |
|
|
1-90 days |
2 |
4 |
3 |
2 |
11 |
|
|
91-365 days |
0 |
0 |
3 |
2 |
5 |
|
|
1< years ≤ 2 |
1 |
1 |
2 |
1 |
5 |
|
|
> 2 years |
0 |
3 |
1 |
0 |
4 |
|
|
TOTAL |
3 |
8 |
9 |
5 |
25 |
408 Dr Marks undertook the same exercise with respect to the placebo arms of the Alzheimer’s studies:
Table 6. Summary of Alzheimer’s studies placebo AEs that occurred off‑drug and were counted by Professor Madigan in his analysis. Results summarise time on drug and time to event after drug discontinuation.
Follow‑up Time Off-Drug Until AE Occurs
|
Days on Drug |
1-90 days |
91-365 days |
1< years≤2 |
> 2 years |
TOTAL |
|
|
1-90 days |
1 |
1 |
1 |
0 |
3 |
|
|
91-365 days |
3 |
0 |
0 |
0 |
3 |
|
|
1< years ≤ 2 |
0 |
0 |
0 |
1 |
1 |
|
|
> 2 years |
0 |
1 |
0 |
0 |
1 |
|
|
TOTAL |
4 |
2 |
1 |
1 |
8 |
409 With respect to the inclusion by Prof Madigan of events which had occurred more than 14 days after discontinuation, Dr Marks said:
Note that for the 25 Vioxx subjects whose CVT event occurred more than 14 days after drug discontinuation, 11 (44%) were on their study drug for no more than three months. In addition, 14 of the events (56%) occurred more than one year after drug discontinuation, with 25% being more than two years after drug discontinuation. Only three Vioxx events (12%) occurred within three months of drug discontinuation. … These results indicate generally little Vioxx exposure in these subjects and resulting CVT events that occurred long after Vioxx exposure ends. There are considerably fewer placebo events, and they generally occurred in subjects with little placebo exposure. Those events, unlike those in the Vioxx group, tended to cluster in the first 90 days after their drug discontinuation. A cardiologist needs to interpret these results and determine if there is any reasonable biological relationship that can be shown between short‑term Vioxx use and long‑term occurrence of CVT events.
Generally with respect to Prof Madigan’s analysis, Dr Marks said:
The end result here is that Professor Madigan has combined numerous categories of events from the Alzheimer’s studies, some non‑standard categories (off‑drug and nonserious) and one inappropriate category (not confirmed by adjudication to be CVT) to arrive at a statistically significant result. Indeed, this fragile [relative risk] quickly drops to that of no risk when these categories are scrutinized. Such results are not considered proof of a documented Vioxx CVT risk.
While under cross‑examination by counsel for the applicant, Dr Marks was not challenged on this evidence.
410 I generally accept Dr Marks’ defence of Merck’s use of confirmed rather than investigator‑reported events in the Alzheimer’s studies. As the respondents pointed out, if the investigator‑reported data in a study show a higher incidence of the event of interest than do the confirmed data, it follows that some events considered to be of interest by investigators were regarded otherwise by the blinded, specialist, adjudicators. The question remains, however: should the data which Merck did use be regarded as compromised, or of little present utility, by reason of the circumstance that events which had occurred more than 14 days after discontinuation were not counted? In the way this issue was debated by the parties, it had two aspects, one procedural and one substantive.
411 The procedural issue related to the matter dealt with in the passage from the evidence of Prof Madigan set out in para 401 above. It was submitted on behalf of the applicant that Merck had originally specified an ITT approach to the collection of data, but subsequently departed from that stated intention. Indeed, in his final written submissions the applicant contended that the terms of Protocol 078 had been amended so as to convert an ITT study to what was effectively an on-drug study. Although not so alleged in terms, the implicit gravamen of the applicant’s submission was that this had been done surreptitiously. The submission related to a passage in the original protocol. In the “Data Analysis” section, under the sub‑heading “Statistical Methods”, the following appeared:
The primary approach for all efficacy and safety analyses will be the “intention‑to‑treat” approach. All patients will be included in the analysis in the treatment group to which they are randomized, regardless of any protocol violation.
The applicant submitted that this passage had been deleted without comment and “the ITT analysis disappeared from the document without a noted deletion”. I do not accept that submission. Amendments were made on 8 May 1998, but there was no deletion of the passage quoted above. The applicant submitted that “[n]o future document mentioned these changes, rendering it impossible for anyone who did not look at the original protocol to know that ITT was prespecified for safety.” The reason that no future document mentioned the changes was that they had not been made. Likewise, the passage in question was in Protocol 091 and 126 from the outset, and remained there subsequently.
412 How did Merck align the use of its “modified ITT” approach with the statement in the passage set out above that the approach for all safety analyses would be the ITT approach as such? Here Dr Reicin made a distinction between the terms of the protocol and the terms of the data analysis plan for each study, focusing specifically on Protocol 078. She considered that the language of the protocol was “ambiguous”. Beneath the contentious passage set out above, the text was divided up according to three sub‑subheadings, namely “Primary Efficacy Analysis”, “Interim Analysis” and “Safety”. Under “Safety”, it was said: “All patients who take study medication will be included in the analysis of safety.” By contrast, in the section on safety the data analysis plan contained the following passage:
Two approaches will be used for tabulation of adverse experiences ... First, a primary analysis will include all adverse experiences occurring when patients are on treatment and up to 14 days following discontinuation of study drug … Second, a summary based on the ITT approach will include all adverse experiences regardless of whether patients were on or off study drug … Each safety subsection will present the on‑drug results followed by the respective ITT results.
Dr Reicin said that it was standard practice at MRL for the data analysis plan to take precedence over the protocol with respect to the statistical analyses that were conducted when analysing the data from a clinical trial.
413 The evidence is not such as would entitle me to decide whether, in some absolute sense, Merck was right or wrong to prefer the data analysis plan over the protocol. However, that was not Dr Reicin’s only procedural justification for the 14‑day cut-off system which was used. Under cross‑examination, it was put to her that it was because of the data analysis plan for Protocol 078 that she took the view that “an on‑drug analysis was preferable.” She responded:
It wasn’t an issue of preferable or not. That was the primary stated analysis for safety and for [cardiovascular] safety it specifically referred to our standard operating procedure which was on drug and within 14 days.
Here Dr Reicin was referring to the CV SOP. Protocols 078 and 091 both pre‑dated the CV SOP (078 was dated 4 December 1997 and 091 was dated 14 October 1998). Merck established the CV SOP specifically for the purpose of providing a consistent, across‑study, basis for monitoring the association between the consumption of Vioxx and the occurrence of CVT events. Such an association was not a pre‑specified concern of the Alzheimer’s protocols as such. Other studies, looked at in isolation, may have taken an approach different to that of those protocols. If, as apparently was contemplated from the outset, there was to be pooling of data across studies, an important consideration would have been consistency in the basis upon which events of interest were recorded. I take the view that it was reasonable of Merck, in relation to the Alzheimer’s studies, to treat the 14‑day approach specified in the CV SOP as primary for the purpose of collecting and analysing CVT events.
414 In deciding whether Vioxx in fact led to an increased risk of CVT disease, the essentially procedural issues which I have just been discussing are of less importance than the answer to the question whether an ITT or an on-drug approach to the collection and analysis of data would have been the better calculated to reveal that risk, if there were one. That is the substantive issue to which I earlier referred. Here there are several threads of evidence which bear upon the subject. There is the recognition by Prof Madigan himself, contained in the passage set out at para 401 above, that the use of an ITT approach may have the effect of diluting safety signals that would otherwise arise from the data. That is because, I infer, data (including the number of patient‑years accumulating in the denominator of the risk fraction) ostensibly generated by the experiences of patients taking the drug which is presumptively harmful would in fact be coming from patients who were acting as though they were taking placebo. Much the same point was made by Dr Reicin:
Although ITT is considered to be a conservative approach when proving efficacy or superiority of the treatment drug, an on‑drug analysis is more conservative when examining safety issues or proving non‑inferiority because off-drug events will tend to dilute any drug effects. International standards in the design of clinical trials support that position: “Although considered conservative for efficacy, ITT analyses are not conservative for analyzing adverse event data where the results will be biased, on average, towards finding no differences between the groups (in other words, a tendency for the groups to be more similar to each other than they should be).” … [Management of Safety Information from Clinical Trials: Report of … Council for International Organizations of Medical Science Working Group VI, Geneva 2005 at 137]. …
415 Next there is the evidence of Dr Marks, set out at paras 407‑408 above, in which he points to some fairly extreme cases of events which would be counted under a true ITT approach. Although one must accept Prof Madigan’s point that to ignore a serious event that occurred on the 15th day after discontinuation might be difficult to justify, nonetheless the line has to be drawn somewhere, and to count an event that occurred a year or more after discontinuation might equally, especially if the patient concerned had been on the drug for a short period only, have the potential to distort the picture conveyed by the study.
416 The biostatisticians also had something to say in their joint report on this subject. They said:
11. We agreed that it is appropriate to conduct per‑protocol (PP) and intention‑to‑treat (ITT) analyses for safety endpoints.
12. We agreed that if there exists no a priori belief that a drug could cause a persistent (ie off‑drug) risk, it is reasonable to perform PP and ITT analyses, with the PP analysis as primary. We agreed that one would expect the PP and ITT analyses to agree and any disagreement would lead to further investigation and analyses.
13. Drs. Madigan and Woodward agree that if there is an a priori belief that a drug could cause a persistent risk, then ITT analyses are primary. Dr. Marks believes both PP and ITT have equal importance.
In the case of Vioxx, the nature of the “a priori belief” that guided Merck in the development of the CV SOP was identified by Dr Reicin as follows:
It is important to remember that our analyses of cardiovascular safety of Vioxx were conducted against the backdrop of the FitzGerald hypothesis: that COX-2 selection decreases prostacyclin … without affecting thromboxane … in the blood vessels, creating an imbalance leading to an increased risk of blood clotting. This hypothesized pharmacological mechanism, if true, would suggest an acute on‑drug effect rather than long‑term risk.
This evidence effectively opens up another question which was considered by the cardiologists, namely, whether the effect of Vioxx in the vasculature (assuming there to be one) was acute only as suggested by Dr Reicin, or endured over the long term, eg by promoting the development of atherosclerosis. I deal with that matter at paras 546‑550 below.
417 Prof Zipes relied on the extended data generated by the APPROVe trial. As I indicate elsewhere in these reasons, Prof Zipes had a view, not shared (in the case of Prof Harper, at least to the same extent) by the other cardiologists, that Vioxx accelerated atherosclerosis. In that sense, its toxic effect might persist well after discontinuation. As Prof Celermajer pointed out, that is not the realm of the FitzGerald hypothesis. Notwithstanding, Prof Zipes said that “the fact that there was continued risk for that one year after stopping Vioxx in the APPROVe data would suggest that some of the changes indeed do remain and the individual is still at risk.”
418 It is apparent that there is no bright line between situations in which an ITT approach and situations in which an on-drug approach will be most informative on matters of safety. Neither is it uncontentious whether an ITT or an on-drug approach should have been regarded as the better suited for a study designed to test the cardiovascular risks associated with the consumption of Vioxx. In this part of my reasons, there is no reason why I should not derive assistance from whatever evidence is available. As it happens, Dr Marks has divided the data in a way that makes it possible to put to one side adverse events that had been reported by investigators but not confirmed on adjudication. When that is done, the relative risk of suffering a CVT event from taking Vioxx rather than placebo is seen to be 1.24 (0.90, 1.69), on an ITT approach.
419 The difference between this figure and the one reported to the FDA in January 2005 (as adjusted by the inclusion of Protocol 126 – 1.05 (0.71, 1.54)) is entirely the result of the inclusion of events that occurred more than 14 days after discontinuation (it will be seen from Dr Marks’ table that the inclusion of non‑serious events in Prof Madigan’s data actually lowered the relative risk figure). Given the direction of this difference, I think it is unlikely that any safety signal would have been “diluted” by the inclusion of events occurring after the 14th day. I think it more likely either that there was in fact something about Vioxx which gave rise to an excess of CVT events over placebo to a strikingly greater extent within this extended period than is observed up to the 14th day after discontinuation, or that, by chance, unrelated circumstances lay behind that excess. Although Profs Zipes and Harper expressed the view that the harmful effect of Vioxx persisted, they did not go to the extent of opining that its relative toxicity actually increased with the passage of time off‑drug. Perhaps regrettably, no party asked the cardiologists to express an opinion about the data referred to by Dr Marks, as set out in para 406 above. Absent such an opinion, I am in no position, guided by the data alone, to conclude that the risk of consuming Vioxx as disclosed by the Alzheimer’s studies was any greater than disclosed in the reports in fact submitted by Merck to the FDA.
420 In his report, Prof Madigan also expressed strong reservations as to the rates of compliance amongst participants in the Alzheimer’s studies. His point was that, if patients were regarded as taking Vioxx when in fact they were not, any pro‑thrombotic tendency in the drug would be concealed, to an extent at least. He said:
Significant compliance problems existed in the Alzheimer’s studies generally and in 078 in particular. Only 10.9 percent of the patients on Vioxx and 16.2 percent of patients on placebo remained on their study drug for the entire study period. Only 61 percent of patients on Vioxx took at least 80 percent of their assigned tablets, and 25.8 percent of these patients took the drug less than 60 percent of the time. The study was prematurely terminated 11 months prior to its scheduled end. By contrast, in VIGOR, 99.4% of Vioxx and 99.1 % of naproxen patients had a compliance rate of at least 75%. In APPROVe, 91.9% of Vioxx and 94.9% of placebo patients had a compliance rate of at least 75%, and only 3.9% of Vioxx and 2.8% of placebo patients recorded a compliance rate of less than 60%. Since noncompliance would generally tend to diminish the effect size associated with the active treatment, the results … [set out in Prof Madigan’s analysis] are all the more striking in the face of such high levels of noncompliance.
421 Dr Marks responded to these criticisms. He said that the ITT approach had been taken for efficacy in the Alzheimer’s studies because they were designed to assess residual drug efficacy for cognition after drug use ended. Thus, patients were treated as still being in a trial even if they had stopped taking the drug. For example, a patient who had discontinued half‑way through the trial would have had a compliance rate of 50%. By contrast, a patient in VIGOR or APPROVe who discontinued half‑way through would have a compliance rate of 100%. Dr Marks said that there was “no problem with the quality of data resulting from the Alzheimer’s studies, … but instead [the quality of the data] simply reflects the difference in measuring compliance in an ITT versus [per‑protocol] analysis”. In his statement in reply, Prof Madigan accepted the force of Dr Marks’ observations, but contended that, while the difference was less pronounced, the compliance rate in Protocol 078 was less than that in APPROVe. In a further statement, Dr Marks set out the compliance rates in APPROVe and in Protocol 078 in a table which, as I understand it, was intended to present the figures on a comparable basis:
Table 11. On‑drug compliance rate comparison for APPROVe and Alzheimer’s protocol 078. “Mean compliance” refers to the average study drug compliance attained across all subjects, and “>75%” refers to the percentage of subjects having at least 75% study drug compliance
------ APPROVe ----- ----- Alzheimer’s 078 -----
Vioxx Placebo Vioxx Placebo
Mean compliance 93.1% 94.7% 86.9% 90.0%
>75 % 91.9% 94.9% 83.1% 88.3%
Dr Marks said:
Although the Alzheimer’s compliance rates are numerically lower than for APPROVe, these compliance rates are still very good for an RCT. Reported results from protocol 078 should not be questioned based on low compliance.
422 Under cross-examination, Prof Madigan conceded that what Dr Marks said about compliance rates was correct. He described the compliance rates in the Alzheimer’s studies as “a bit lower” than those in VIGOR. I do not think that the utility of the Alzheimer’s data should be regarded as compromised by reason of low levels of compliance amongst patients taking Vioxx.
VICTOR
423 The applicant submitted that the results of the VICTOR study also showed “a strong signal for CVT risk”. That study was, along with APPROVe and ViP, part of Protocol 203. It was terminated early when Vioxx was withdrawn from the market. In the study, 2434 patients with colorectal cancer were randomised to Vioxx 25 mg daily or placebo. It had been intended to enrol 3500 patients for two years’, and a further 3500 patients for five years’, follow‑up. Prof Celermajer said that “the study’s eventual number of patient years follow‑up was less than 10% of that planned”. To the time when the study was terminated, the occurrence of CVT events yielded a relative risk figure (for taking Vioxx) of 2.66 (1.03, 6.86). It seems that a disproportionate number of the patients in the Vioxx arm were diabetics and, after adjusting for that circumstance, the figure was 2.41 (0.93, 6.26); p = 0.07. Prof Celermajer took the view that the study was “not particularly informative”. Counsel for the respondents submitted that VICTOR did not “replicate the APPROVe results”. I consider, however, that counsel for the applicant were closer to the mark when they submitted in reply that “VICTOR showed a RR of CVT events of about 2.5 which is either statistically significant, or just shy of statistical significance, depending on the analysis used”. Although the adjusted relative risk figure of 2.41 was not statistically significant, the p figure shows that there would only be 7 chances in 100 that one would be in error to exclude the hypothesis that there was no difference in the incidence of CVT events as between those who took Vioxx and those who did not. Allowing for the statistical limitations on use of the data involved here, although it might be strictly true that those data do not “replicate” APPROVe, they are conspicuously consistent with it.
Other meta analyses
424 The applicant next relied on two meta‑analyses of RCTs which, he submitted, reinforced the conclusion that the consumption of Vioxx increased CVT risks and the risk of myocardial infarction. The first was that of Kearney (2006). In his report, Prof Celermajer provided the following summary of this research:
The authors performed a meta‑analysis of published and unpublished tabular data from any randomised trial comparing COX-2 inhibitor use to placebo or traditional NSAIDs; where trials were ≥ 4 weeks duration and vascular event data were available. In the placebo comparisons, they found a [relative risk] of 1.42 (95% CI 1.13‑1.78) for serious vascular events, for any coxib versus placebo. They found no evidence of differences in the [relative risk] associated with different coxibs. The overall result was chiefly driven by a difference in the relative risk of acute [myocardial infarction], at 1.86 (95% CI 1.33‑2.59) with no significant difference in the rates of other vascular events in subjects on COX-2 inhibitors …
The applicant relied on the relative risk figures of 1.42 for serious vascular events and 1.86 for myocardial infarction as providing support for his case on risk.
425 Profs Celermajer and Vaughan offered no criticism of Kearney. Indeed, they appeared to regard it as a good study (subject, of course to the caveats conventionally expressed in relation to meta‑analyses by comparison, for example, with RCTs). In the joint report of the cardiologists, they relied upon Kearney for their opinion that the available data disclosed no significant difference between coxibs with respect to cardiovascular outcomes; and for their opinion that that data did not show that Vioxx predisposed to thrombotic stroke. Speaking of Kearney, Prof Vaughan said:
I think the Kearney analysis does the most thorough job of giving us a number with regard to the absolute risk related to the administration of a COX-2 inhibitor and that analysis gives us a number of about three events per 1,000 people allocated to a COX-2 inhibitor per year.
I think what Prof Vaughan meant when he referred to the “absolute risk” was the increased risk arising from the taking of Vioxx rather than placebo. An examination of the Kearney article shows that the 3 per 1000 per year figure to which Prof Vaughan referred was the excess of events for those on coxibs by comparison with those on placebo. The absolute figures were, for coxibs, 12 per 1000 per year and, for placebo, 9 per 1000 per year. It may also be relevant to note that these figures were for “vascular events” as defined in the study, namely, myocardial infarction, stroke and vascular death.
426 Setting the background for their study, Kearney and colleagues said:
The accumulating evidence suggests that selective COX-2 inhibitors are associated with an increased risk of vascular events, but several important questions remain unanswered. Firstly, what is the magnitude of any excess risk of myocardial infarction, stroke, and vascular mortality? Secondly, is the excess risk of vascular events dose related, and is the size of this risk different in people who are also taking aspirin (which chiefly inhibits COX-1 at low doses)? Thirdly, are traditional NSAIDs (which also inhibit COX-2) associated with an increased risk of vascular events?
The endpoints of interest were serious vascular events, defined as fatal or non‑fatal myocardial infarction, fatal or non‑fatal stroke and vascular death (including death from myocardial infarction or stroke). It is not clear whether haemorrhagic stroke was included, but no party suggested that the results were not useful on that account. For the sake of simplicity, I shall treat Kearney as dealing with CVT events.
427 Kearney and colleagues presented their placebo‑controlled data in the following form:
428 Although the authors stated the relative risk numerically only when giving the composite results for the five coxibs being studied, it is clear from the numbers that that risk figure, in each case, was strongly influenced by the rofecoxib and celecoxib data. Using the data in the Kearney paper, Prof Woodward calculated the relative risk of myocardial infarction from ingesting rofecoxib rather than placebo. It was 1.74 (1.11, 2.72). He undertook no such calculation for vascular events generally. It was implicit in the cases of both parties, however, that I should accept the composite figure of 1.42 (1.13, 1.78) as broadly applicable to rofecoxib. I consider that the Kearney study provides firm, and relatively uncontroversial, support for the applicant’s case on risk.
429 The other meta‑analysis relied on by the applicant was that of Juni (2004). The authors examined the results of 18 RCTs, in which Vioxx had been compared with placebo, with naproxen and with non‑naproxen NSAIDs. Their endpoint of interest was myocardial infarction. They examined the results by reference to the dose of Vioxx being taken. Their broad finding, taking all comparators and all doses into account, was that the relative risk of suffering a myocardial infarction from taking Vioxx rather than a comparator was 2.24 (1.24, 4.02). Their finding in relation to situations in which Vioxx was taken at a daily dose of 25 mg (all comparators) was that the relative risk was 1.37 (0.52, 3.61). When the comparator was placebo (all doses), the figure was 1.04 (0.34, 3.12).
430 The overall risk figure derived by Juni and colleagues was criticised by the respondents as being skewed upwards by the gravitational pull of VIGOR (ie because of the size of the sample in that trial). They made their point by reference to the following figure in the published article:
The size of the black squares is an indication of the size of the patient samples in the various studies. Of this figure, Prof Celermajer said:
So if one looks at figure 2, until the very biggest black square which is VIGOR, all the dots lie near unity. When VIGOR is introduced, the square moves to the right of unity and when you follow it vertically downwards, the black square moves back towards unity again. So the Juni meta‑analysis is driven entirely by the VIGOR result, which … is flawed because of the comparator dose of naproxen and the high dose of Vioxx.
Dr Marks made much the same point. The respondents’ submission was that, if the contention that the VIGOR results were largely the result of the cardioprotective work of naproxen is reasonably open, Juni went no further than to beg the question. It did not, at a statistically significant level, establish that Vioxx increased the risk of myocardial infarction.
431 The respondents’ experts criticised Juni also for having omitted from the meta‑analysis trials the results of which might have been thought to favour Vioxx. Most obviously in this regard, the Juni analysis did not include the results of the Alzheimer’s studies. Prof Woodward defended the authors on this aspect, pointing out that the timing of the publication did not permit them to obtain the safety data from the FDA for those studies. I accept that defence so far as it goes, but, in present context, the exclusion from a meta‑analysis of a significant body of data where the comparator was placebo must compromise the utility of the research. I also accept that the inclusion of a large trial in which naproxen was the comparator is not obviously calculated to address the questions left unanswered by VIGOR. In their final written submissions, counsel for the applicant ignored these contentious aspects of Juni. They contented themselves with drawing attention to the results as such. I accept the respondents’ reservations as to this study. I consider that its utility as an indicator of the tendency of Vioxx to cause myocardial infarction is limited.
432 The respondents relied on oral evidence given by Prof Celermajer, the purport of which was to show that the relative risk (point estimate) of about 2 – observed in relation to CVT events in APPROVe – was atypical. Indeed, it was submitted that this figure lay within the 95% confidence interval for the relative risk of taking Vioxx rather than placebo in only a minority of studies. Prof Celermajer drew together what he described as “the best placebo‑controlled data” to put against the relative risk figures yielded by APPROVe. The first study which he mentioned was Konstam (2001) which, as I have indicated, contained the same data as were forwarded to the FDA on 8 January 2001. For APTC endpoints, the relative risk of taking Vioxx as against placebo was 0.84 (0.5, 1.4). The next study was the Juni (2004) meta‑analysis. There, the relative risk for myocardial infarction (although not expressly so limited by Prof Celermajer in his evidence) of taking Vioxx rather than placebo was found to be 1.04 (0.34, 3.12). Prof Celermajer next referred to the Alzheimer’s disease studies, in which regard he understood that the biostatisticians had agreed that the relative risk of encountering a CVT event if taking Vioxx rather than placebo was found to be (on‑drug) 0.97 (0.7, 1.4) or (ITT) 1.24 (0.9, 1.7). The next study was APPROVe itself, in which, for CVT events, the risk of taking Vioxx rather than placebo was said to be 1.9 (1.2, 3.1). The next was the Kearney (2006) meta‑analysis, where the relative risk in the case of serious vascular events was 1.4 (1.1, 1.8). The final study referred to by Prof Celermajer was another meta‑analysis, this time relating only to myocardial infarctions, published as Chen (2007), wherein the relative risk of taking rofecoxib rather than placebo was found to be 1.38 (0.87, 2.19).
433 Profs Harper and Zipes each responded to Prof Celermajer’s analysis. Prof Harper said:
I have not heard of statisticians agreeing that when you have a placebo controlled trial that is the gold standard then lumping that together with lots of meta‑analyses of small studies which in themselves have no power to show an end point. Comparing that data to a placebo controlled randomised study is not appropriate and I do not [resile] from my statement that I think Vioxx at a dose of 25 milligrams has a risk factor of 2. Perhaps even higher. If you look at the APPROVe study … the total thrombotic adverse events, the risk ratio was 1.92 but if you looked at cardio events the risk ratio quoted in that study is actually 2.98 [sic – the correct figure was 2.80].
Prof Celermajer agreed with Prof Harper that a placebo‑controlled randomised clinical trial was the “sine qua non” of evidence, but added that there was a lot of evidence available, and that to throw it all out without regard for how it “contextualises” APPROVe was to risk losing much valuable information. Prof Zipes’ response to Prof Celermajer was:
I find [Professor Celermajer’s] analysis not acceptable. It is combining oranges and apples and whatever else. However, if one were to accept that – which I don’t but if one were to – my conclusion is that Professor Celermajer agrees that there is risk associated with Vioxx. It’s just a question of how much.
434 I shall consider the studies described by Prof Celermajer as involving the best placebo-controlled data in turn. If one is attempting to find the most informative data, I cannot understand why the pooled analysis of January 2001 (which was the basis for the Konstam article) should be used in preference to the latest such data forwarded by Merck to the FDA (in March 2004). Neither can I understand why the APTC endpoint would be regarded as more valuable in the context of the question of the pro‑thrombotic tendency of Vioxx than the CVT endpoint (although I accept, of course, that the January 2001 data contained only the APTC endpoint). If the March 2004 data and if the CVT endpoint are used, the relative risk of taking Vioxx rather than placebo is seen to be 1.26 (0.89, 1.78). Having said that, Prof Celermajer’s point, that a point estimate of 2 does not lie within this confidence interval, remains good (albeit that the interval does overlap with that observed in APPROVe).
435 As set out above, Prof Celermajer’s relative risk figure does represent that reported by Juni (2004). It relates to myocardial infarction only. Being confined to the placebo comparison, the utility of the figure is not compromised in the way of Juni’s overall risk figure as discussed in para 431 above. However, although the point estimate is close to unity (and for that reason the figure is unhelpful to the applicant), the confidence interval is wide enough to encompass each of the presently relevant point estimates in APPROVe (ie 1.92 for CVT, 2.53 for myocardial infarction and 2.80 for cardiac events generally).
436 I do not understand the biostatisticians in the present case to have agreed that the correct relative risk figures to be derived from Merck’s Alzheimer’s studies were those stated by Prof Celermajer. Merck’s figure – based on confirmed CVT events occurring up to the 14th day after discontinuation – was 1.03 (0.70, 1.53). Prof Madigan’s figure (which I have declined to accept) – based upon all investigator‑reported events at any time – was 1.5 (1.1, 2.0). Prof Madigan also supplied a figure which incorporated confirmed events for the “on‑drug” period and investigator‑reported events thereafter: it was 1.27 (0.91, 1.79). Aside from the investigator‑reported analysis of Prof Madigan, it is true that the confidence intervals stated here do not incorporate a risk figure of 2 or thereabouts.
437 Prof Celermajer’s figure for the relative risk of a CVT event as shown in Kearney (2006) is correct; and he is correct to say that a relative risk of 2 does not fall within the confidence interval (which was 1.13, 1.78). It might be noted, however, that the myocardial infarction risk figure reported by Kearney was 1.86 (1.33, 2.59), where the confidence interval does cover the point estimate of the relative risk for myocardial infarction yielded by the APPROVe data (2.53), although only just.
438 Curiously perhaps, very little reference was made in the cases of the parties to Chen (2007). In the evidence here under discussion, Prof Celermajer relied on it. He correctly reported the relative risk figure for myocardial infarction where the comparator was placebo. Prof Woodward relied on it in his witness statement in reply. He was evidently impressed with the quality of the data from Chen, based as they were on clinical trials rather than observational studies. He said that the data supported the view that the consumption of Vioxx was associated with myocardial infarction. In the final submissions on behalf of the applicant, there was no reference to this evidence or to Chen. The respondents mentioned Chen, but with little emphasis. For myocardial infarctions, it presents a very different picture from that presented by APPROVe (although, as Prof Woodward said, being “indicative of an effect”).
439 There was a further meta‑analysis upon which Prof Woodward relied: McGettigan (2006). This study involved an examination of the data from 17 case‑controlled and 6 cohort studies, of which 9 and 2 respectively involved a comparison between use of Vioxx and non‑use. Cardiovascular events – predominantly myocardial infarction – were the endpoints of interest. The relative risk of taking Vioxx (all doses) as observed in those 11 studies was 1.31 (1.15, 1.59). Neither party relied on, or referred to, McGettigan in final submissions. Neither was it mentioned in any of the oral evidence. There was something of a technical issue played out in the witness statements of Prof Woodward and Dr Marks which related to this study, Dr Marks contending that, in relation to retrospective studies, it was accepted in the scientific community that nothing less than a statistically significant relative risk of 3 should be regarded as evidence of causation. Only two of the comparisons tabulated by McGettigan (and, as it happens, none of those that go to make up the relative risk figure referred to earlier in this paragraph) satisfied that condition. However, Prof Woodward pointed out that it was a “misconception [to contend] that studies which are, themselves, inaccurate can … not make a valuable contribution to a meta‑analysis”. This study, therefore, supports both the applicant’s case that Vioxx causes CVT disease and Prof Celermajer’s contention that the relative risk as against placebo is somewhat less than that shown in APPROVe.
Other studies
440 The applicant relied also on a study of another coxib, valdecoxib, published as Nussmeier (2005). In that study, 1671 patients who had undergone “CABG” (coronary artery bypass grafting, pronounced “cabbage”) were randomised into three arms – the first to be given paracoxib (the analog of valdecoxib) intravenously for three days followed by (oral) valdecoxib for seven days, and the third to be given placebo for 10 days. All were taking prophylactic aspirin. The relative risk of encountering a cardiovascular event (including, but not limited to, myocardial infarction) was found to be 3.7 (1.0, 13.5). Although Prof Zipes referred to this study as evidence in support of the FitzGerald hypothesis, Prof Celermajer said:
The situation of cardiopulmonary bypass is a highly unusual one in that a patient’s blood is circulated through an extra‑corporeal circuit of plastic tubing and this results in significant perturbations of the coagulation and haemostatic systems. Furthermore, the coronary arteries are physically manipulated and occasionally subjected to surgical trauma, as well as the heart/lung bypass circuit, often resulting in a systemic pro‑inflammatory response. For these reasons, conclusions made about COX-2 inhibitors for the relief of post‑operative pain in patients who have had open heart surgery are certainly valid, but only for that specific clinical situation.
For those reasons, and because the study was not concerned with Vioxx as such, I am disposed to think that the work of Nussmeier and colleagues is of little assistance to the applicant in the present case.
441 Prof Zipes referred also to the APC study, to which I have referred at para 343 above. He relied upon the statistically significant relative risk of taking celecoxib rather than placebo with respect to cardiovascular events. Profs Celermajer and Vaughan, however, regarded this study as at least potentially corroborative of their view, expressed primarily in relation to APPROVe, that colon polyp patients may be an atypically susceptible group. Dr Reicin referred to the APC study as tending to indicate that such cardiovascular risk as might arise from the consumption of Vioxx was by way of a “class effect” in which all coxibs were implicated. As with the post‑CABG study, the APC study did not involve Vioxx as such. Since it showed a statistically significant increase in the risk of taking celecoxib rather than placebo, I am prepared to regard it as providing a measure of support for the applicant’s case, but it must also be recognised that, to the extent that the respondents have a point about the susceptibility of colon polyp patients, the study likewise gives them a measure of support for treating the results of APPROVe as not generalisable.
The mortality data
442 In my consideration of the data from the various Merck trials and studies, and from published materials, I have followed the parties by focusing upon the events – generally APTC and/or CVT – reported in those sources. Additionally, the parties drew my attention to a particular category of data which, it seems, are commonly if not invariably collected in RCTs and similar studies. They are the mortality data, that is, a record of the deaths occurring amongst participating patients. To a statistician, deaths occurring for any reason will be at least potentially of interest, since the pattern of them may show an association. So too will it be of interest to know the pattern of deaths occurring as between patients taking the drug of interest and patients taking the comparator. To a clinician, of course, the cause and circumstances of each death will be of interest, since they may aid in the determination of a biological, rather than a merely statistical, association between the therapy and the death.
443 In the trials conducted in accordance with the CV SOP, Merck’s “modified ITT” approach required a death to be counted if it had occurred while the patient was on study therapy or within 14 days after discontinuation or, where the death was to be ascribed to an adverse experience of interest, whenever it occurred if the adverse experience happened, or began, on therapy or within those 14 days. However, it seems that all deaths were sent for adjudication, even those that were not to be counted in the analysis. This meant that, in general, it was possible to identify how many participants in a particular study had died as a result of, for example, a CVT event, whether the event occurred either before or after the 14th day after discontinuation.
444 Although mortality data were available in respect of all the Merck studies to which the court was referred, the applicant relied only on the data which came from the Alzheimer’s studies. As a result, those data, and how the deaths in those studies might have been otherwise reported and analysed, came to occupy an atypically conspicuous place in the forensic controversy. For that reason, I shall commence with them.
445 Although Merck forwarded to the FDA all the raw mortality data from the three Alzheimer’s protocols, it carried out statistical analyses only with respect to Protocols 078 and 091 (probably because they were the only studies that had run their course according to the original specification). In a Combined Safety Analysis for those protocols sent to the FDA on 17 December 2003, Merck said that “[t]here were 41 (3.84%) deaths from all causes in the rofecoxib group and 23 (2.14%) in the placebo group.” The relative risk (of taking Vioxx) was 2.23 (1.33, 3.73). Of these data, the author of the analysis said:
The difference in deaths was due to increases in the “Injury, Poisoning and Procedural Complications,” “Infections and Infestations,” and “Cardiac Disorders” categories. … There was no apparent pattern in the specific cause of death to suggest an increased risk with rofecoxib. Overall, the number of patients who died during the trials was small and the findings should be interpreted with caution due to the fact that no statistical multiplicity adjustment was performed.
Dealing with the subject of CVT mortality, the author said:
There were 11 (l.03%) patients in the rofecoxib arm whose deaths were adjudicated as due to a thrombotic cardiovascular event and 5 (0.47%) in the placebo arm. The patient‑year adjusted incidence rates were 0.66 and 0.26 per 100 patient‑years for the rofecoxib and placebo arms, respectively.
The relative risk (of CVT death from taking Vioxx) was said to be 2.56 (0.89, 7.35).
446 My attention was not drawn to any such combined analysis which included also the data from Protocol 126. For the record, however, I note from the Clinical Study Report, sent to the FDA on 22 July 2002, that there was one confirmed CVT death in the Vioxx arm and two such deaths in the placebo arm. If these figures are added to those referred to in the Combined Safety Analysis for Protocols 078 and 091, it will be seen that there were 12 CVT deaths in the Vioxx arms of the three studies and seven in the placebo arms.
447 Prof Madigan analysed the CVT mortality data yielded by the Alzheimer’s studies, but he did so in two ways that differed from Merck’s analysis of December 2003. First, he included the data from Protocol 126. Secondly, he used true ITT data, since all deaths had been sent for adjudication. Although he did not set out the crude numbers on which he based his calculation, his evidence was that, by reference to data available in June 2003, the relative risk of CVT death from taking Vioxx rather than placebo was 3.49 (1.48, 9.68). Dr Marks did not challenge these figures in his statement in reply to Prof Madigan. Although Prof Madigan was cross‑examined on this aspect, there was no suggestion that his figures were wrong, or that his analysis was misconceived.
448 Of the mortality data from the Alzheimer’s studies, Dr Reicin said:
In the Spring of 2001, medical statisticians at Merck conducted an interim analysis of data from the Alzheimer’s studies. An all‑cause mortality analysis revealed that there were statistically significantly more deaths in the Vioxx group than in the placebo group. ... Cardiovascular mortality was not the key driver behind the difference in overall mortality. Cardiovascular mortality showed a small numerical imbalance that did not reach statistical significance. MRL found that there were numerically more accidental deaths and deaths related to infection on Vioxx than on placebo. Two examples include a patient that was electrocuted and an Alzheimer’s patient that ingested a bromide tablet that significantly burned his esophagus that led to infections that ultimately caused his death.
Dr Reicin made no comment on what was shown by the final figures (as distinct from the “interim analysis”) from the Alzheimer’s studies. Although she submitted a statement in reply which dealt extensively with the report of Prof Madigan, she did not challenge his mortality figures; neither did she refer to the final mortality data of December 2003. Clearly her reference to deaths arising from irrelevant causes is no answer to Prof Madigan’s analysis of the confirmed CVT deaths. While it may be accepted that deaths from non‑CVT causes “drove” the overall mortality figures in one or other, or both, of the arms of the studies, the more relevant question for present purposes relates to the difference in the risk of CVT death as between the two arms.
449 In their final written submissions, the respondents referred to Prof Madigan’s CVT mortality figures from the Alzheimer’s studies. They submitted that those figures relied on off‑drug events (which I am prepared to assume they did). They cited Dr Reicin’s evidence, set out above, that cardiovascular mortality “showed a small numerical imbalance that did not reach statistical significance”, describing it as being based on “a more detailed analysis”. That was not a satisfactory response to Prof Madigan’s point. Dr Reicin’s evidence clearly does not bespeak “a more detailed analysis” than that undertaken by Prof Madigan. The respondents relied on Dr Marks’ evidence as to cardiovascular events generally which occurred long after discontinuation of study treatment. I have accepted that evidence, but neither Dr Marks nor any other expert applied it in the specific context of confirmed CVT deaths.
450 It is possible that something might have been made of the different patterns of CVT mortality as between Protocol 078 and Protocol 091 with respect to ITT versus on‑drug data collection. The Clinical Study Report for Protocol 091 noted that there was only one death that did not result from an adverse experience that began while the patient concerned was receiving therapy or within 14 days after discontinuation. That patient, an 81 year‑old female from the Vioxx arm, took Vioxx for 35 days, discontinued, and, 148 days later, experienced an adverse event which led to death. By contrast, it appears from the Clinical Study Report for Protocol 078 that, of the total of 24 deaths classified as “cardiac disorders” (a grouping which I accept is not co‑extensive with CVT disorders) (18 in the Vioxx arm and 6 in the placebo arm), 10 (9 in the Vioxx arm and 1 in the placebo arm) resulted from adverse experiences occurring after the 14th day after discontinuation. Prof Zipes referred to follow-up work which had been undertaken by Merck in relation to Protocol 078. Dealing with deaths generally (ie not confining himself to CVT deaths), Prof Zipes said:
Of note however, the sponsor continued to accrue data on a significant number of subjects after discontinuation of the study, i.e., 356 in the Vioxx arm and 307 in placebo. Over a median follow up of 29 and 20 weeks respectively, 17 deaths occurred in the Vioxx arm versus only 5 in placebo. The authors do not offer statistical analysis, although it is clearly very significant. Indeed, this is a disturbing signal, consistent with long‑term toxic effects.
This evidence, which was given in Prof Zipes’ original report, was referred to neither in the answering reports of Profs Celermajer and Vaughan nor in the joint cardiology session. Such evidence as the cardiologists might have been able to give on the differences in the ITT data (compared to the on‑drug data) as between Protocols 078 and 091, and how they might have related to the different study designs involved, could well have thrown light on the potential for Vioxx, if taken over an extended period, to produce harmful effects over the long term. As I have said, however, the issue was not explored.
451 Doubtless because they rejected the ITT approach as such, the respondents made no attempt to reconcile their mortality figures with Prof Madigan’s. The applicant too contented himself with relying on Prof Madigan’s figures (although, as I have said, by reference to investigator‑reported events). Indeed, his final submissions contained no reference to the analysis of CVT confirmed deaths undertaken by Prof Madigan. The parties’ cases do, in these respects, pass each other like ships in the night. It may be that some or all of the CVT confirmed deaths resulting from events which occurred more than 14 days after discontinuation so occurred a very long time thereafter, and did so in circumstances in which it might have been clinically adjudged that they had nothing to do with the treatment received in the studies (such as, possibly, the death of the patient referred to in the previous paragraph). However, the court has the benefit of no such analysis of individual events as Dr Marks provided in relation to adverse events generally.
452 In due course I shall consider whether it was unreasonable for Merck to have taken the 14‑day approach to CVT events leading to death in the Alzheimer’s studies. I am here concerned not with that question but with the question whether the data from the studies, viewed in the most informative way, in fact implied the existence of CVT risk from taking Vioxx. Here I have a relative risk figure supplied by Merck to the FDA of 2.56 (0.89, 7.35). In broad terms, whether or not the events from Protocol 126 are included, there were about double the number of relevant deaths in the Vioxx arms as in the placebo arms. To the extent that a relative risk calculation is available (ie for Protocol 078 and 091 only), the difference is not significant. But the direction of the signal is unambiguous. Prof Madigan’s analysis tells us that, if CVT events which occurred more than 14 days after discontinuation and which led to death are also counted, the relative risk is substantial and significant: 3.49 (1.48, 9.68). If these mortality data were looked at in isolation from those of the Alzheimer’s studies generally, they would have to be regarded as providing some support for the applicant’s case on risk.
453 However – and here I turn to the other studies and trials of which evidence was given – I can think of no reason why they should be looked at in isolation. Commencing with VIGOR, there were 22 deaths (0.5%) in the Vioxx arm and 15 deaths (0.4%) in the naproxen arm. Prof Celermajer said that the overall mortality rate was similar in the two arms, and he was not challenged on that evidence. These overall figures would not appear to provide a source of comfort for Merck, but, within them, the most common form of death in the Vioxx arm (four deaths) was pneumonia, while the most common forms of death in the naproxen arm (four deaths each) were pneumonia and myocardial infarction. Perhaps the most interesting statistic is that there were two deaths from myocardial infarction in the Vioxx arm and (as already noted) four deaths from that cause in the naproxen arm.
454 With respect to Merck’s arthritis studies, Dr Reicin said:
The Phase IIb/III osteoarthritis studies showed statistically significantly fewer deaths in the Vioxx group than in the NSAID group (diclofenac, ibuprofen, and nabumetone). This difference was driven by more cardiovascular deaths in the NSAID group than in the Vioxx group.
Dr Reicin was not challenged on that evidence.
455 In the APPROVe trial, there were ten deaths in each arm that either occurred or arose from adverse events which occurred while the patients concerned were on study therapy or within 14 days of discontinuation. Of these, six in the Vioxx arm and four in the placebo arm arose from thrombotic causes. There were a further 21 deaths which occurred, or arose from adverse events which occurred, after the 14th day but during the anticipated three‑year study periods for the patients concerned: 13 in the Vioxx arm and eight in the placebo arm. Of these, three in each arm were thrombotic.
456 The only other analysis to which it is useful to refer in the present context is Kearney (2006). It will be seen that the authors’ Figure 1, reproduced in para 427 above, includes an analysis of “vascular death”. Although I recognise that that category is not co‑extensive with CVT death, it provides some kind of indication that the excess of deaths in the Vioxx arms of the studies there examined was only slightly above unity at the point estimate and not nearly statistically significant.
457 In the course of the cardiology concurrent session, Prof Celermajer said:
[T]here is absolutely no evidence that Vioxx predisposes to fatal cardiac events. … The signal here is for non‑fatal myocardial infarction. There is no signal for fatal heart disease in any of the Vioxx trials, either in meta‑analysis [or] in the APPROVe study.
Despite the fact that this evidence (which was given in the course of an exchange with the court) was not later challenged, I am not prepared to accept it at face value, as the respondents invited me to do in their final written submissions. First, as Prof Zipes immediately pointed out, the Alzheimer’s data revealed a statistically significant excess of overall mortality in the Vioxx arm. It is true that Prof Celermajer was referring to cardiovascular death specifically, as to which there was no (at least uncontroversial) statistically significant excess in the Vioxx arms of the Alzheimer’s studies, but I consider that it did these (non‑significant) data less than justice for Prof Celermajer to have said that “there is no signal for fatal heart disease in any of the Vioxx trials”. Secondly, Prof Celermajer’s comment that “the signal here is for non‑fatal myocardial infarction” is not quite correct either. Under the CV SOP, fatal and non‑fatal myocardial infarctions were counted. The “signal” in VIGOR, and in APPROVe, related to myocardial infarction, whether or not death resulted. Prof Celermajer would have been more correct to say “the signal here is for myocardial infarction generally, not for fatal myocardial infarction as such”.
458 Notwithstanding those qualifications, it is as clear as may be that the only evidence of any conspicuous association between the consumption of Vioxx and CVT death resides in the data from the Alzheimer’s studies. That evidence is only slightly less conspicuous when those data are looked at through Merck’s 14‑day prism than when Prof Madigan’s ITT approach is taken. Ultimately, I consider that the difference between those two approaches is less weighty for present purposes than the difference between the Alzheimer’s mortality data, on the one hand, and the like data from other studies and trials, on the other hand. Taking everything into account, I could not conclude that there was most likely something about Vioxx that led to death as a result of a CVT event, as distinct from leading to the event itself. Neither could I conclude that, if the generality of the evidence is not such as to persuade me on the probabilities that the consumption of Vioxx increased the risk of CVT events, the mortality data would make any material difference.
The dose relationship
459 The question whether the dose at which Vioxx was ingested had an impact on the risk of CVT events is presently relevant for two reasons. First, the dose relationship is the sixth Bradford Hill criterion. No party suggested that the observed presence of such a relationship ought not to be taken into account in addressing questions of risk. Secondly, if there is such a relationship, it would go part of the way to explaining the results of VIGOR. In that trial, it will be recalled, the dose of Vioxx taken was 50 mg daily, twice the recommended therapeutic dose.
460 In their joint report, the cardiologists agreed that the data were “variable, consistent with a dose effect but not conclusive”. They went beyond that, however, in their separate opinions. Profs Zipes and Harper held the view that the adverse effect of Vioxx was dose‑dependent. They relied upon the study published as Graham (2005), where the authors distinguished between patients taking 25 mg or less and patients taking more than 25 mg. Possibly because this was an observational study of the experiences of contributors to a large health care organisation in California, there was no placebo arm as such. There was, however, an arm described as “remote use” of NSAIDs – where use had ceased more than 60 days before the index date. The relative risk figures – for acute myocardial infarction taking Vioxx rather than remote use of any NSAID – were 1.23 (0.89, 1.71) for the standard dose of Vioxx and 3.00 (1.09, 8.31) for the higher dose. Prof Harper accepted that, ideally, one would not rely on observational studies, but the Graham research was one place where he had been able to find a statistical treatment of the relative risk of taking Vioxx at different doses. Profs Harper and Zipes also contrasted the results of APPROVe (where the dose was 25 mg) with those of VIGOR (where the dose was 50 mg). To them, this comparison (even allowing for some modest effect of naproxen in VIGOR) tended to point to a dose relationship.
461 When looking at the sixth Bradford Hill criterion, it was not in the respondents’ interests to submit that there was a dose relationship, but they did submit, and the experts called by them expressed the opinion, that the VIGOR results were at least partly to be explained by the high dose of Vioxx involved there. In his report, Prof Celermajer drew attention to the APC trial (Solomon 2005), where the relative risk of CVT events from taking celecoxib rather than placebo (for colon polyp patients) was 2.3 (0.9, 5.5) at a dose of 200 mg twice daily, but 3.4 (1.4, 7.8) at a dose of 400 mg twice daily. He said:
Therefore it is plausible that doubling the dose of rofecoxib above the recommended daily dose may have been a major factor in the observed excess CV risk of rofecoxib, when compared with naproxen in the VIGOR study.
Prof Celermajer referred also to the observational study of Ray (Oct 2002), where the relative risk of acute myocardial infarction or fatal coronary heart disease (for current users of Vioxx) was, at doses of or less than 25 mg, 1.03 (0.78, 1.35) and, at doses greater than 25 mg, 1.70 (0.98, 2.95); and (for persons new to the drug during the study) it was, at doses of or less than 25 mg, 1.02 (0.76, 1.37) and, at doses greater than 25 mg, 1.93 (1.09, 3.43). To these data might be added the data of McGettigan (2006), who found that the relative risk of CVT events (mostly myocardial infarction) from taking Vioxx rather than placebo was 1.33 (1.00, 1.79) at doses of or less than 25 mg, but 2.19 (1.64, 2.91) at higher doses. Prof Woodward relied on these data to conclude that the relationship between the consumption of Vioxx and myocardial infarction was dose‑dependent.
462 The data just discussed appear to give a fairly consistent picture of a dose relationship. However, the two major meta‑analyses of RCTs discussed in the evidence – Juni (2004) and Kearney (2006) – are less helpful than one might have hoped. The relative risks yielded by the Juni data were:
Dose Relative risk
12.5 mg 2.71 (0.99, 7.44)
25 mg 1.37 (0.52, 3.61)
50 mg 2.83 (1.24, 6.43)
The endpoint of interest was myocardial infarction, and the authors commented that they “recorded no evidence to support the notion that rofecoxib’s cardiovascular toxicity is dose‑dependent”. They did not remark upon the very odd‑looking data set out above. At a dose of 50 mg those data were heavily influenced by the inclusion of the VIGOR results: for about three‑quarters of the patients who took that dose, the comparator was naproxen. For these and other reasons canvassed above, perhaps the Juni study ought to be approached with reserve. No such qualification applies to Kearney, but, unhappily for present purposes, there were too few vascular events amongst patients taking higher or lower doses of Vioxx than 25 mg to enable an evaluation of dose‑dependence. There was a weak trend towards greater risk with higher doses of celecoxib, but that trend was driven by the results of one study (Solomon 2005).
463 Although, as I have said, the Juni and Kearney studies do not confirm the picture conveyed by the other studies to which I have referred, neither do they confound it. Taking everything into account, I am persuaded that there is sufficient to justify the conclusion of Profs Zipes and Harper that such an association as there was between taking Vioxx and encountering CVT events, including myocardial infarction, was dependent, at least to an extent, on dose. This is, of course, an intermediate conclusion based on the epidemiology, and does not take biological factors into account. However, I consider that there is sufficient in the evidence to infer dose‑dependence within the framework of the Bradford Hill criteria.
Consistency of association across studies
464 To assess consistency across a broad range of studies, I have found it convenient to set out a summary of the relative risk figures referred to above in tabular form as follows:
|
|
CVT Events |
Myocardial Infarction |
||||
|
Lower |
Point |
Upper |
Lower |
Point |
Upper |
|
|
VIGOR |
1.39 |
2.38 |
4.00 |
1.72 |
5.00 |
14.29 |
|
APPROVe |
1.19 |
1.92 |
3.11 |
1.11 |
2.53 |
6.28 |
|
APPROVe extend (Baron) |
1.18 |
1.70 |
2.46 |
1.09 |
1.94 |
3.34 |
|
Merck pooled 22 March 04 |
|
|
|
|
|
|
|
* Rheumatoid arthritis |
0.43 |
3.57 |
164.23 |
|
|
|
|
* Osteoarthritis |
0.97 |
2.84 |
8.38 |
|
|
|
|
* Alzheimer’s disease |
0.70 |
1.03 |
1.53 |
|
|
|
|
* All |
0.89 |
1.26 |
1.78 |
|
|
|
|
Madigan RA/OA |
1.30 |
3.10 |
9.20 |
|
|
|
|
Madigan AD, CV death |
1.48 |
3.49 |
9.68 |
|
|
|
|
VICTOR adjusted |
0.93 |
2.41 |
6.26 |
|
|
|
|
Juni (all doses) |
|
|
|
0.34 |
1.04 |
3.12 |
|
Kearney (all coxibs) |
1.13 |
1.42 |
1.78 |
1.33 |
1.86 |
2.59 |
|
Kearney (Vioxx) per Woodward |
|
|
|
1.11 |
1.74 |
2.72 |
|
McGettigan (mostly MI) |
1.15 |
1.35 |
1.59 |
|
|
|
|
Chen |
|
|
|
0.87 |
1.38 |
2.19 |
465 Several things should be said about this table. First, save for VIGOR (where the comparator was naproxen) the comparator in each group of data in the table was placebo. Secondly, I have not included the results reported by Konstam (2001), Reicin (2002) or Weir (2003) as those results are effectively comprehended, and superseded, by the pooled analysis of March 2004. Thirdly, where the data were controversial as between the parties in this case, I have prepared the table in accordance with the findings I have made earlier in these reasons. Fourthly, I have set out the figures in a way that places the point estimate between the lower and upper 95% confidence limits, both to assist in a ready understanding of the table and to emphasise that the statistically correct statement conveyed by a particular set of figures is that the true relative risk would lie, in 95% of the cases, between those limits. Fifthly, I have included the McGettigan data in the CVT column, but, as noted, those data were predominantly made up of myocardial infarctions.
466 As I have said, APPROVe was the primary trial upon which the applicant relied. Are the results of the other studies consistent with it? Commencing with VIGOR, there are two particular circumstances which, I accept, most probably contributed to the higher (and, in the case of myocardial infarction, much higher) rate of events in that trial. The first is that the comparator was naproxen. Although the experts did not express it this way, to recognise that the beneficial effect of naproxen was to an extent responsible for the VIGOR results appears to introduce greater, not less, harmony with the APPROVe results. In the case of myocardial infarctions, if naproxen (taken twice daily consistently) is assumed to reduce the risk by about a third, and possibly by up to a half (ie on the basis that the benefit of naproxen in VIGOR may have been similar to that found in Watson (2002) and Kimmel (2004)), one might suppose that up to about a half of the 5.0 relative risk figure in VIGOR was attributable to the work of naproxen. The second circumstance is the 50 mg dose which was used in the VIGOR trial. Although it is impossible to quantify the dose effect, for reasons given above I accept that there most probably was one.
467 Because of the uncertainties involved, it would, in my view, be going too far to say that VIGOR provided support for the actual risk figures yielded by APPROVe. None of the expert witnesses proposed any scientific means by which the risk figures of VIGOR might be adjusted to allow for naproxen and dose. I would not presume to undertake that task. However, those two factors together clearly had the potential to explain the difference between the data of the two trials. In this sense, I am persuaded that the conclusion, presented primarily by APPROVe, that Vioxx was associated with a higher incidence of CVT disease, and of myocardial infarction in particular, is consistent with the results of VIGOR.
468 Turning to the other data set out in the table above, the first impression one gets is that there is nothing like a uniform picture of the extent of the relative risk involved in consuming Vioxx rather than placebo. Such picture as emerges is patchy. However, certain things can be said. First, wherever a statistically significant relative risk is shown, it is a risk of consuming Vioxx rather than placebo. Secondly, even where the figures are not statistically significant, the point estimate lies above unity. Thirdly, the non‑Merck meta‑analysis which attracted the most conspicuous support in the submissions of the respondents – Kearney – yielded a statistically‑significant positive risk of taking coxibs both for CVT events and for myocardial infarction. As Prof Woodward demonstrated, the same may be said for Vioxx as such in relation to myocardial infarction. Fourthly, as against these considerations, the Merck Alzheimer’s disease results were not significant and yielded a point estimate very close to unity. So too are the Juni myocardial infarction figures equivocal, but they were based on early data and are not, in my view, sufficient to qualify the conclusion otherwise proper to be reached from the data in this table. That conclusion, in my view, is that the results of the studies referred to in the table are broadly consistent with APPROVe, at least to the extent of showing a greater risk of CVT events and of myocardial infarction if Vioxx rather than placebo is taken.
469 As problematic as it obviously is, it is also necessary that I attempt at least a broad quantification of the extent to which the studies referred to, taken together, suggest that these risks were increased for patients taking Vioxx. From the perspective of public health authorities and manufacturers such as Merck, it may be enough to conclude that there was an increased risk. Such a conclusion will usually be sufficient to warrant the taking of some kind of corrective action. However, the exercise with which the court is presently concerned involves the ex‑post determination of the cause of an event, or series of events, which has happened. Here the task will be advanced to no small degree if it is possible to identify the extent of the increased risk associated with the activity or substance of interest.
470 Although the simplest approach to this task is to take the point estimate of the relative risk figures yielded at statistically significant levels by various studies, it is important to bear in mind what such figures convey. By stating the point estimate, one does not identify a unique figure which, at the 95% confidence level, represents the relative risk. As made clear earlier in these reasons, the point estimate is only the “best estimate” of the true relative risk. That estimate is not made with 95% confidence. Rather, the statement that can be made with 95% confidence is that the true relative risk lies within the confidence intervals which invariably accompany the point estimate as reported. Subject to that caveat, and being conscious of the limitations involved, it is not inappropriate to use the point estimate as a general indicator of relative risk. The parties and their experts did so in this case, and I propose to do likewise.
471 It was the point estimate for the relative risk figure yielded by the APPROVe results that formed the basis for the conclusion of Profs Zipes and Harper that the risk of encountering a CVT event approximately doubled when a person took Vioxx. As I understood their position, they would say that the risk of myocardial infarction would increase by a factor of about 2.5, also because of the results of APPROVe. Profs Celermajer and Vaughan disagreed. In Prof Celermajer’s reading of the studies taken as a whole, the more typical relative risk figure was seen to be about 1.4 (for CVT events) (see para 432 above).
472 In my consideration of this question, I should remember that, although the question must be decided on the balance of probabilities, it is a question of central importance in this case. Here the words of Dixon J in Briginshaw v Briginshaw (1938) 60 CLR 336, 361 have direct application:
The truth is that, when the law requires the proof of any fact, the tribunal must feel an actual persuasion of its occurrence or existence before it can be found. It cannot be found as a result of mere mechanical comparison of probabilities independently of any belief in its reality.
473 In this forensic context, I consider that the results of the various studies, trials and analyses referred to above warrant a number of observations. First, notwithstanding the technical quality of APPROVe, for reasons expressed above, the generalisability of its results must be regarded as a matter of some uncertainty. Secondly, the extended analysis of the APPROVe results, to which I was invited to have regard by the applicant, show relative risk figures, both for CVT events and for myocardial infarction, which are lower, and within tighter confidence intervals, than the original figures (notwithstanding that the lower limits of the confidence intervals remained effectively unchanged). Thirdly, the one meta‑analysis on which I was invited to rely by both sides, and of which neither was critical, yielded a relative risk figure for CVT events (within fairly tight confidence limits) in line with Prof Celermajer’s figure of 1.4, and for myocardial infarction which was broadly consistent with that yielded by the APPROVe follow‑up data. It is true that the Kearney study related to a number of different coxibs, but it may be noted that Prof Woodward’s analysis of the Vioxx‑only myocardial infraction data revealed a relative risk figure that was lower (albeit only slightly) than that yielded by the data generally. Fourthly, despite their limitations, there is nothing in the McGettigan data (which were not drawn exclusively from RCTs) or in the Chen data (which did not yield a statistically significant result) which would tend to contradict any of the observations made above.
474 Turning to the other available data, while I am persuaded that the VIGOR results play an appropriate role in supporting the conclusion that the consumption of Vioxx increased the risk of CVT events and myocardial infarction, I do not think they are useful in the quantification of that risk. I have held that naproxen and dose both most probably contributed to the results, but any attempt on my part to place a figure on the extent of that contribution would be altogether too hazardous, particularly when I have rejected the applicant’s evidence that the naproxen effect was no more than about 2%. I am prepared to say that the VIGOR results are not inconsistent with those of APPROVe and the APPROVe follow‑up study, but I could not go to the length of finding that they supported a quantification, say, of something more than 2 rather than of something less than 2.
475 It is Prof Madigan’s combined arthritis figures that make a striking contrast with those of APPROVe and the meta‑analyses. What Prof Madigan did, by pooling, was to give statistical significance to the relative risk data of the separate Merck osteoarthritis and rheumatoid arthritis studies. In the case of the original osteoarthritis data, the relative risk figure was very nearly significant and, subject to that qualification, the point estimate for CVT events was substantially greater than the corresponding figure yielded by the other studies to which I have referred. I would say the same about Prof Madigan’s relative risk figure for cardiovascular death in patients covered by the Alzheimer’s studies. However, Prof Madigan’s figures related only to CVT events as a group, and tell us nothing (at least directly) about myocardial infarction. Also, I am here concerned to estimate, so far as possible, the extent to which the consumption of Vioxx increased the relative risk of encountering a CVT event as a general proposition, rather than (at this stage at least) to make such an estimate with respect to patients suffering from a particular condition. In that setting, I can see no reason not to recognise that the (non‑fatal) data yielded by the Alzheimer’s disease studies, although non‑significant, conveyed no message of risk, or not to combine those data with those from the arthritis studies.
476 In the result, I am persuaded that, taken as a whole, the data referred to warrant generalisation that, over a population, the consumption of Vioxx increased the risk of CVT events by a factor of about 1.5, and of myocardial infarction by a factor of about 2. I make these findings because of the allegations in the applicant’s pleading and because of the questions which I am required to address. Their practical utility, however, must be understood to be subject to two qualifications. First, they are based on the statistical results of the trials and studies referred to. They take no account of issues of mechanism, to which I shall turn next. And secondly, they relate, as I have said, to people generally. The data may have a rather different utility when the circumstances of a particular person, suffering a particular condition, are required to be considered.
The FitzGerald hypothesis
477 Turning to the third Bradford Hill criterion – the existence of a plausible biological explanation – the applicant relied primarily on the FitzGerald hypothesis. It will be convenient to commence by considering the nature of CVT disease. In terms with which the other cardiologists did not disagree, Prof Vaughan laid out the basic steps in the development of CVT disease as follows:
1) injury to the arterial wall; 2) the adherence and migration into the vessel wall of monocytes (a certain type of cell); 3) uptake of oxidized LDL (low‑density, or “bad” cholesterol) into the arterial wall; 4) progression of lipid accumulation within the atherosclerotic plaque and development of a necrotic, lipid core; 5) destabilization and thinning of the plaque’s fibrous cap overlying the necrotic, lipid core; 6) rupture or erosion of the fibrous cap exposing the highly prothrombotic contents of the plaque to the circulating blood; and 7) thrombosis and resulting ischemia.
478 Prof Zipes explained that there were “roughly four … main stages” of atherosclerosis:
The first stage for the development of atherosclerosis is that of endothelial injury, or damage to the inner cellular lining of the blood vessel wall (by hypertension, shear stress, toxins like nicotine, diabetes, etc.). The damaged inner lining attracts the adherence of white blood cells to its surface… The second stage is one of fatty streak formation. The LDL cholesterol (“bad” cholesterol) penetrates the endothelium … and accumulates in the vessel wall. When this LDL is oxidized as a result of its penetration into the arterial wall … it sets up an inflammatory reaction in the endothelium, attracting more white blood cells that ingest the oxidized LDL. These white blood cells called monocytes become “foam cells” after they ingest the LDL, and are the hallmark of the fatty streak … These cells also elaborate a host of inflammatory mediators … that promote inflammation in the fatty streak, regulate the direction, amplitude and duration of the immune process, and contribute to the progression of the lesion and formation of a plaque. The third stage is one of fibrous cap formation, which is probably the first step in the development of a complex lesion that can cause an acute thrombosis … that can close off an artery. This fibrous cap prevents the lipid core that has accumulated beneath it from contacting the blood pool. But the cap is susceptible to damage from activated white blood cells that secrete enzymes which weaken the collagen in the cap. Inflammation, occurring for reasons that are poorly understood, appears to play an important role in the activation of white blood cells that later undermine the fibrous cap. The final stage is the development of a complicated atheroma, which typically occurs when rupture of the weakened fibrous cap causes the blood to come into contact with the lipid core, causing the formation of a thrombus. Physical disruption of the atherosclerotic plaque, due to fracture of the fibrous cap or superficial erosion of the intima, commonly causes acute thrombosis. Thrombus formation occludes the blood vessel, interrupting blood flow to the perfused organ, in this case the heart. If the thrombus dissolves spontaneously, it may cause a healing response that thickens the fibrous cap, making it bulge into the lumen of the artery and further restricting blood flow by the atheroma. If the thrombus persists and completely occludes the vessel, the downstream myocardium dies due to lack of blood flow, which is the definition of [myocardial infarction].
479 It is the seventh step in Prof Vaughan’s sequence, or the fourth stage referred to by Prof Zipes, where the imbalance hypothesized by Dr FitzGerald and his colleagues is said to have a biological impact, favouring the development of thrombotic material in the vasculature.
480 In the joint report of the cardiologists, Profs Zipes and Harper said that the FitzGerald hypothesis was biologically plausible and provided a reasonable explanation for the observed association between the consumption of Vioxx and adverse cardiovascular events. They would apply that conclusion to all COX-2 inhibitors, but added that Vioxx at a dose of 25 mg daily had a more powerful effect on COX-2 inhibition than Celebrex at a dose of 200 mg daily, and therefore carried a greater risk of adverse cardiovascular events at those doses (ie consistently with the FitzGerald hypothesis). By contrast, Profs Celermajer and Vaughan said that they were “not convinced that the FitzGerald hypothesis is biologically relevant to an understanding of the pathophysiology of plaque rupture and thrombosis.” Prof Vaughan said that there was nothing to suggest that the mechanisms proposed in the FitzGerald hypothesis were unique to selective COX-2 inhibition. He said that all COX inhibitors, with the exception of low-dose aspirin and high-dose naproxen, were statistically similar with respect to cardiovascular risk.
481 I have explained the origins, and broadly the content, of the FitzGerald hypothesis at paras 43‑47 above. In final written submissions on his behalf, the applicant put it as follows:
The basic premise of the FitzGerald hypothesis is that selective inhibition of COX-2, while sparing COX-1, upsets the critical balance between prostacyclin and thromboxane, thereby increasing the likelihood of thrombosis.
That premise was, however, articulated in a rather more sophisticated way in the evidence of the cardiologists called by the applicant. In his witness statement, Prof Zipes relied upon the following passage in Antman (2005):
COX-1 is the only isoenzyme expressed in platelets; endothelial cells express both COX-1 and COX-2. In the normal artery, the balance between PGI2 and TXA2 production favors PGI2 and inhibition of platelet‑dependent thrombus formation. In the atherosclerotic artery, both PGI2 and TXA2 production is increased, owing in part to increased platelet activation with compensatory PGI2 formation via both COX-1 and COX-2 in endothelial cells; the net effect is an imbalance favoring TXA2 production and platelet‑dependent thrombus formation. Low‑dose aspirin selectively impairs COX-1‑mediated TXA2 production in platelets restoring the net antithrombotic balance. COXIB use suppresses COX-2 dependent PGI2 production in endothelial cells, which has only a marginal effect on the net antithrombotic balance owing to the importance of COX-1 as a source of PGI2 in the normal state. In the setting of atherosclerosis, however, COX-2 plays a greater role as a source of PGI2 and more TXA2 is produced; thus, inhibiting COX-2 in this setting has a more profound effect on prostanoid balance, favoring TXA2 production and promoting platelet dependent thrombosis.
Likewise, Prof Harper said:
The drug Vioxx is a selective inhibitor of the [COX-2] enzyme. Inhibition of this enzyme in atherosclerotic arteries has been shown to adversely alter the balance between the production of prosta‑cyclin … which is a protective substance secreted in the lining cells of blood vessels and thromboxane A2 … which is a substance secreted by platelets. TxA2 is a potent vaso‑constricting compound that is highly active in aggregating platelets and acts to promote clot formation. … In contrast, PGI2 is a vaso‑dilator substance which causes dilatation of blood vessels and directly antagonises the constricting and procoagulant effect of TxA2. Vioxx unfavourably alters the balance between these two agents by blocking the production of PGI2 thereby enhancing the effect of TxA2 thus increasing the risk of clot formation on an athero‑sclerotic plaque.
Save for standing as explanations of the mechanism hypothesised by Dr FitzGerald, the significance of these statements is that they situated the scientific debate with which the court is presently concerned in the context of the atherosclerotic vasculature.
482 The cardiologists called by the respondents did not accept the FitzGerald hypothesis as a plausible explanation for the relatively greater number of CVT events amongst patients taking Vioxx observed in some trials and studies. Prof Celermajer said that, while he originally considered that the hypothesis “had some potentially attractive features”, later publications had “cast significant doubt on its validity”. He said that there were problems with the hypothesis and its applicability to humans, namely:
(a) The hypothesis reduced a highly complex system to an overly simplistic paradigm.
(b) Because prostacyclin acted locally, and was not a circulating hormone, the presence of the urinary metabolite was, or at least might well be, evidence only of prostacyclin production in the kidney and urinary tract.
(c) Numerous studies had shown that the selective inhibition of COX-2 – or, more accurately, the sparing of COX-1 – did not, as would have been expected, lead to an increase in the urinary levels of thromboxane metabolites.
(d) In the studies that had been conducted, there was no evidence that the supposedly adverse effects of COX-2 inhibition were neutralized by the inhibition of aspirin.
(e) A 1979 study found that even a 90% decrease of prostacyclin release by endothelial cells still left enough prostacyclin to prevent platelet aggregation in humans in vivo.
(f) In a 1981 article, Dr FitzGerald himself suggested that the vasculature did not produce enough prostacyclin for it to be a systemic antiplatelet agent.
(g) There was doubt over whether endothelial cells produced prostacyclin via the COX-2 pathway.
(h) Contrary to what one would expect if the FitzGerald hypothesis were valid (an early sign of the increased incidence of CVT events in patients taking Vioxx), in the APPROVe trial the risk of CVT events appeared to emerge only after about 18 months. The data on the time‑relationships involved in the emergence of risk were not consistent between trials, but they did not provide support for the hypothesis.
(i) There were multiple, mutually‑compensating, systems which regulated thrombosis and bleeding in the vasculature. It was unlikely that perturbation of any single system would not result in some counter‑balancing compensatory change of another system.
(j) Recent studies involving other coxibs – lumiracoxib in 2004 and etoricoxib in 2006 – did not reveal significant differences between groups, on an ITT analysis.
(k) Recent studies which showed a possible increase in cardiovascular risk in the case of patients taking non‑selective NSAIDs called into question the “unbalanced inhibition” aspect of the FitzGerald hypothesis.
Prof Celermajer found items (d) and (h) on this list most important in raising doubts about the FitzGerald hypothesis.
483 Prof Vaughan said that there were “numerous unresolved questions” which limited the interpretation that could be placed on the findings out of which the FitzGerald hypothesis arose. In his report, he continued:
For example, prostacyclin is generally viewed as an autocoid, meaning that it is generated and acts locally. At this time, I am not aware of any studies that have ever been done to define the relevance or magnitude, if any, of COX-2 dependent prostacyclin production in the human coronary circulation. Importantly, there is no evidence to suggest that COX-2 is in fact present in normal human arterial vasculature. Indeed, numerous studies indicate that human arteries exclusively generate COX-1. Furthermore, the studies that suggested that COX-2 inhibition reduced systemic prostacyclin production were based on reductions in the measurement of a stable prostacyclin metabolite in the urine. The precise location of the tissue responsible for this effect and altered prostacyclin synthesis has not been determined. Studies in humans suggest that COX-2 inhibitors do not, however, affect prostacyclin production in the human vasculature … Furthermore, it has been shown that COX-2 inhibition has no effect on endothelial vasodilator function in adults … Taken together, the data from experimental and preclinical studies have thus far failed to generate a consistent picture or understanding of the potential impact that COX-2 inhibitors might or might not have on the progression of human cardiovascular disease.
484 Against this background of contention, it is convenient to turn to the case advanced by the respondents in opposition to the hypothesis. They proposed that there were seven “necessary key assumptions that must apply in both healthy and diseased vasculature for the FitzGerald hypothesis to be operative”. They were:
1. Compensatory systems in the vasculature are insufficient to overcome the prothrombotic effect caused by an imbalance between prostacyclin and thromboxane.
2. Reductions in the prostacyclin urinary metabolite (PGI‑M) correspond to actual reductions in prostacyclin in the vasculature.
3. COX-2 is present in the endothelium.
4. COX-2 rather than COX-1 makes endothelial prostacyclin.
5. A partial reduction in prostacyclin will have a clinical effect.
6. COX-2 inhibition has no effect on thromboxane levels.
7. The balance between prostacyclin and thromboxane is critical for preventing thrombosis.
I am not at all satisfied that each of these points is apt to be described as an “assumption”. As I understand it, the FitzGerald hypothesis is not based on assumptions. It takes as its starting point observed outcomes from pharmacological studies, and hypothesises a biological process consistent with those outcomes. However that may be, these points were used by counsel for the respondents in the cardiology concurrent evidence session to seek to obtain a degree of scientific consensus for the implicit proposition that the FitzGerald hypothesis was in effect unsound. As will be apparent, that endeavour was largely unsuccessful, but the respondents’ points will nonetheless provide a convenient framework within which to assess the strength of the applicant’s case to the extent that it relies on the hypothesis. I shall, however, deal with these points in what I consider to be a logical sequence, rather than in the order in which they are set out above.
485 I take first the second point, namely, that the FitzGerald hypothesis requires that “reductions in prostacyclin urinary metabolite (PGI‑M) correspond to reductions in prostacyclin in the vasculature”. As it seems to me, this was in the nature of a pharmacological premise used in the interpretation of the data on which the hypothesis was based. Profs Zipes and Harper agreed with the proposition that “if the FitzGerald … hypothesis is to be proved to be correct one has to assume that the reduction in the prostacyclin metabolites in the urine necessarily correspond to reductions in prostacyclin in the arterial vasculature”. They also agreed that “the measurement of urinary metabolite, … is a systemic rather than a local measurement”, that “the FitzGerald studies of themselves don’t point to the reduction in prostacyclin in the arterial vasculature” and that the mechanism proposed by the hypothesis was “one possibility but … not demonstrated by the studies”.
486 The respondents then moved to the proposition that the reduction in the urinary metabolite observed in the studies on which the hypothesis was based was probably not sourced in the vasculature (item (b) on Prof Celermajer’s list if problems). However, they so proposed on the strength of studies from which the operation or non‑operation of COX-2 in the endothelium might be inferred. That aspect was more directly the concern of the third and fourth of the respondents’ points, to which I shall turn next. Directly on the question of the source of the metabolite observed in the original studies on which the FitzGerald hypothesis was based, it seemed to be common ground that no direct evidence was available.
487 Turning next to the third of the respondents’ points, namely, that COX-2 is present in the endothelium, all the cardiologists agreed that that circumstance was a necessary precondition to the validity of the hypothesis. There was, however, vigorous disagreement as between Profs Zipes and Harper, on the one hand, and Profs Celermajer and Vaughan, on the other hand, as to whether COX-2 was in fact present in the endothelium. In expressing their respective opinions, the cardiologists referred to studies from which the presence or absence of endothelial COX-2 might be inferred, at which point the debate (inevitably as it seemed to me) dealt also with the fourth point, namely, whether COX-2 rather than COX-1 makes endothelial prostacyclin, and under what circumstances. These points address the problem which was item (g) on Prof Celermajer’s list.
488 Prof Vaughan dealt with the subject at the level of the normal vasculature. I have set out (at para 309 above) what he said on the matter in his original report. He developed this evidence in his second witness statement:
However, the report generally cited to support the proposition that the COX-2 enzyme is present in normal vasculature used cultured endothelial cells. … Furthermore, it is highly debatable that endothelial cells derived from umbilical cords actually display the functional or biological properties of endothelial cells in the human systemic arterial or coronary vasculature. It is well known that endothelial cells functionally transform under the artificial conditions of life outside the body in tissue culture. A more valid and informative way of looking at protein expression and localization in endothelial cells utilizes immunohistochemistry staining of fresh, intact human vasculature. Importantly, these studies demonstrate that COX-2 is not normally present in blood vessels .…
489 As I understood Prof Zipes, however, he regarded it as something of a distraction to focus upon the healthy vasculature. In the healthy, he said, “… [prostacyclin] is not likely to be increased and, therefore, not likely to play an important vasodilator role, so COX-2 inhibition would not be expected to have a major inhibitory effect ….” Indeed, it was as good as common ground between the cardiologists that it was the constitutive enzyme COX-1 that synthesises prostacyclin in the normal, healthy, state. Rather, Prof Zipes asserted that the inducible enzyme COX-2 synthesised endothelial prostacyclin under inflammatory conditions: see the extract from his original report at para 481 above.
490 In short, Prof Zipes contended that, in an atherosclerotic or like inflammatory state, the production of prostacyclin is in effect up‑regulated by COX-2. He referred to several studies in support of that conclusion, most notably the one published as McAdam (2005). Since this article was relied on also by the respondents (in respects to which I shall refer presently), it became a significant focus of debate in the cardiology concurrent session. In the study, smokers and non‑smokers received rofecoxib 25 mg twice daily or placebo for one week each in random sequence. The synthesis of systemic thromboxane and prostacyclin was assessed by an analysis of their respective urinary metabolites. Serum TXB2 was measured as an indicator of platelet COX-1 activity. It was found that, at baseline, the excretion of the urinary metabolite of prostacyclin was significantly higher in smokers than in non‑smokers, that both were reduced in the group taking rofecoxib, and that the reduction in the smoking group was significantly greater than the reduction in the non‑smoking group. The study confirmed previous work that thromboxane synthesis was greater in smokers than in non‑smokers. In non‑smokers, there was no significant difference in the synthesis of thromboxane as between the placebo arm and the rofecoxib arm. However, in smokers, the administration of rofecoxib produced a 21% reduction in the synthesis of thromboxane as measured by the urinary metabolite, while there was no reduction in the placebo group. The administration of rofecoxib produced no significant difference in the levels of serum TXB2. In their article in Circulation, McAdam and colleagues expressed the following conclusion:
In conclusion, prostacyclin biosynthesis in cigarette smokers was found to be derived largely from COX-2. This major reduction in prostacyclin biosynthesis during COX-2 inhibition in smokers, however, was not accompanied by an increase in the level of the urinary metabolite of TXA2. Indeed, rofecoxib lowered TXA2 metabolite levels in smokers to a greater extent than in nonsmokers, which indicates that TXA2 biosynthesis via COX-2 is increased in cigarette smokers.
491 In his first witness statement, Prof Zipes said of the McAdam study that it supported –
… the notion that [prostacyclin] is predominantly derived from endothelial COX-2 because it was increased by smoking, which would not be expected to affect renal [prostacyclin] production, but also suggesting that some non‑platelet TXA2 is also COX-2 derived.
In concurrent evidence, Prof Zipes said that the study demonstrated –
… [t]hat the smokers and non‑smokers had very different responses, that the inflammatory response from the smoking is interpreted to have increased prostacyclin and hence, their elevated excretion following COX-2 inhibition and [the authors draw] the conclusion from that that it more likely than not is from the endothelium and it has been induced by COX-2.
This final proposition – as to the locus of the prostacyclin seen to be elevated in smokers – was not accepted by the respondents. When asked to identify the passages in the article which supported his position, Prof Zipes was offered by counsel for the respondents, and accepted, the opportunity to consider the matter over the course of an adjournment. By the time counsel returned to the point, the McAdam article had been extensively canvassed by the cardiologists in connection with another of the respondents’ points. Prof Zipes was then asked to identify the passages to which he had originally intended to refer, and he did so. He was not, at that stage, reminded of the terms of the question, in relation to which he had intended to search the article. Although he did respond by identifying certain passages, they did not, at least on my understanding of them, establish the proposition that the elevated production of prostacyclin observed in smokers had an endothelial source.
492 Prof Vaughan dealt also with the presently controversial issues in the context of the unhealthy vasculature, in which respect he said that “[i]nvestigators have directly measured prostacyclin metabolite levels in the blood of patients with atherosclerosis and failed to see a reduction in patients taking a COX-2 selective inhibitor”. Here Prof Vaughan relied on two studies, only one of which was the subject of any observation by Prof Zipes. That one was published as Kuklinska (2005). Prof Zipes agreed with the proposition that this study, involving a cohort of patients with a clinically obvious cardiovascular disease, demonstrated that a low dose of rofecoxib did not diminish prostacyclin synthesis. However, it appeared that patients in both arms of the study were receiving statins. Prof Zipes’ comment on the study was that the FitzGerald hypothesis had “nothing to do with statins” and that the study did not, therefore, “in any way disprove that hypothesis”. He said that statins were anti‑inflammatory, and “for all we know may preserve prostacyclin”. It also appeared that the dose of rofecoxib being taken was 12.5 mg/day, which Prof Harper described as “a very low dose”. Prof Zipes argued that it was “an insufficient dose to have reduced the prostacyclin metabolism particularly when combined with a statin”.
493 Turning to Prof Celermajer, he said in his report that there was doubt over whether endothelial cells produced prostacyclin via the COX-2 pathway. He said that animal studies had implicated primarily COX-1, and that certain in vitro experiments had found no evidence of COX-2-dependent prostacyclin production by endothelial cells. He referred to a study which had identified the expression of COX-2 in cultured endothelial cells stimulated by being placed under shear stress conditions for 24-48 hours (Topper 1996), but added that more prolonged shear stress (for 7 days) was not associated with an increased expression of COX-2 (Dekker 2002). He observed that there was a difference between isolating and proliferating endothelial cells in a culture dish and the behaviour of such cells in the intact organism. In that respect, Prof Celermajer said that the expression of COX-2 had not been found to be expressed in the endothelial cells of a normal umbilical vein, but that expression of the enzyme appeared only during cell culture.
494 Prof Zipes responded by referring to studies (Hla 1992; Jones 1993) in which “the human nucleic acid sequence of COX-2 was cloned from unstimulated endothelial cells, reflecting the unstimulated expression COX-2”. He said that COX-2 had also been found in atherosclerotic lesions, and mentioned a study in which the expression of COX-2 had been found in human and mouse vessel walls, and in which it was stated: “The failure of some studies to report COX-2 expression in endothelial cells ex vivo (outside the body) may reflect the particular experimental circumstances and/or discordance between the offset kinetics of flow‑induced gene expression and the time of sample preparation.” Prof Celermajer distinguished the studies relied upon by Prof Zipes, in various ways, as being inapplicable to the question whether COX-2 was present in the endothelium. He concluded (in his oral evidence) that “the totality of evidence provided by Professor Zipes in his report does not demonstrate COX-2 protein in relevant human arteries but it suggests that the prostacyclin in the vessels is thereby coming from COX-1”.
495 Ultimately, there was no satisfactory resolution of the disagreement between Prof Zipes, on the one hand, and Profs Celermajer and Vaughan, on the other hand, with respect to the questions whether COX-2 is present in the endothelium and, if so, whether it was responsible for the synthesis of prostacyclin in an atherosclerotic situation. I should not be surprised by that circumstance. The cardiologists’ disagreement lies at the centre of the scientific controversy with which this proceeding has been concerned. The results of Protocol 023 were unexpected for the very reason that it had not been previously thought that COX-2 was present in the endothelium: see the views of Dr Morrison and of the authors of Catella-Lawson (1999) set out at paras 44 and 46 above respectively. Dr Fitzgerald and his colleagues thereafter hypothesised that COX-2 was present in the endothelium, and that the inhibition of it would alter the prostacyclin – thromboxane balance with pro-thrombotic consequences. However, to the extent that the synthesis of prostacyclin is seen to be up‑regulated (inferentially by COX-2) in an atherosclerotic or other inflammatory situation, it does not appear to have been scientifically established that that prostacyclin has an endothelial source. On the other hand, to the extent that the respondents’ endeavour was to disprove (forensically) the FitzGerald hypothesis by establishing the falsity of the third and fourth points proposed by them to be inherent in the hypothesis, I would have to find that they have not done so. Prof Zipes seemed to have a response – which I could not find to be illegitimate – to each of the pieces of scientific evidence upon which Profs Celermajer and Vaughan relied, whether it be that that evidence related only to healthy subjects, that the study in question involved subjects who were taking statins, or similar. If the FitzGerald hypothesis is otherwise to be regarded as providing a plausible biological explanation for the results of the APPROVe study, I am not persuaded that it should be disqualified by the established fact that COX-2 is not present in the atherosclerotic endothelium.
496 The respondents made another point which might be conveniently mentioned at this stage. It relates to Prof Zipes’ disclaimer, in effect, of any claim to the relevance of the healthy vasculature. Profs Celermajer and Vaughan responded by pointing out that the studies on which the FitzGerald hypothesis was originally based involved healthy subjects. As Prof Celermajer put it: “[T]he FitzGerald hypothesis was derived from healthy individuals. I just note that because both Professors Zipes and Harper resile from healthy individual data but that is the data on which the FitzGerald hypothesis is based.” Prof Vaughan referred to the McAdam study of celecoxib, which –
… was performed in healthy individuals and indeed, COX-2 inhibition reduced the levels of the prostacyclin metabolite. This was the whole basis of the hypothesis using healthy people.
In his written submissions in reply, the applicant contended that, in the context of Protocol 023 (Catella‑Lawson 1999), “healthy” should not be regarded as synonymous with “without atherosclerosis”. However, during the examination of the cardiologists by counsel for the respondents in concurrent evidence, counsel for the applicant interjected to make a specific point about whether Protocol 023 and the companion study on celecoxib involved healthy adults. Profs Zipes’ and Harper’s attention was drawn to the point, and they agreed that they did. Counsel for the applicant did not subsequently invite the cardiologists to infer that the study might have involved subjects with atherosclerosis.
497 Because the applicant’s experts effectively limited their reliance on the FitzGerald hypothesis to atherosclerotic or other inflammatory situations, the court is spared the task of addressing the question whether the studies of healthy adults upon which the respondents relied, and which were not consistent with the proposition that COX-2 mediated the synthesis of prostacyclin in the endothelium, compromised the hypothesis. On the other hand, the fact that the studies out of which the hypothesis grew involved healthy subjects only does seem substantially to limit the value of the hypothesis itself in the setting of an atherosclerotic vasculature. The position to which the applicant attached himself was that endorsed by Profs Zipes and Harper, namely, that the FitzGerald hypothesis identified a scientifically plausible explanation for the more frequent occurrence of cardiovascular thrombotic events amongst those taking Vioxx in such a setting, whether or not the experimental facts upon which the hypothesis was originally based were seen to be replicated.
498 I propose next to consider the sixth of the respondents’ points. Here they proposed that the FitzGerald hypothesis required that “COX-2 inhibition has no effect on platelet thromboxane”. In the course of their concurrent evidence, counsel for the respondents had each of the cardiologists agree with that proposition. It became apparent from what followed, however, that the proposition was not understood in the same sense by all present. Prof Zipes made it clear that he understood the proposition to be a reference to the hypothesis itself, rather than to any “assumption” or precondition necessary for its validity. So understood, the proposition was that, conformably with the hypothesis as identified by him, the inhibition of COX-2 would reduce the levels of prostacyclin in the vasculature, while leaving levels of thromboxane unaffected. To confound that proposition, counsel for the respondents first drew Prof Zipes’ attention to so much of McAdam (2005) as showed that, in smokers, the urinary metabolite of thromboxane was “significantly reduced” by taking rofecoxib (ie the 21% reduction referred to above). Prof Zipes agreed that the study did show that. In their written submissions, the respondents contended that this reduction “demonstrates that COX-2 can also produce thromboxane”, which was said to be “inconsistent with the FitzGerald hypothesis”. However, Prof Zipes was not induced to any such concession, and neither Prof Celermajer nor Prof Vaughan so asserted.
499 The scientific purport of this sixth point came to the fore only when Prof Celermajer contributed to the discussion. As will appear, what was apparently intended by the respondents was to propose that, if the FitzGerald hypothesis were sound, one would actually see an increase in the production of thromboxane after the administration of rofecoxib (as suggested by item (c) on Prof Celermajer’s list of problems); or, put differently, that the hypothesis was contradicted by the absence of any such increase in the studies upon which it was based. Prof Celermajer explained the biological basis of that proposal. He explained how, if the mechanism proposed by the hypothesis were empirically valid, one would, inescapably, observe an increase in thromboxane upon the inhibition of COX-2. He said that platelet activation and aggregation were mutually positively reinforcing. An active platelet was more likely to aggregate with other active platelets. In turn that released thromboxane, which brought other platelets in and activated them to aggregate as well. He said that prostacyclin was a specific chemical entity that bound to a receptor on the surface of the platelet to render it quiescent. The loss of that “brake”, as Prof Celermajer put it, should allow the platelets to be active, and to aggregate, on a larger scale than when the brake was on, and if so, one would expect to see an increase in the production of thromboxane.
500 Agreeing with Prof Celermajer, Prof Vaughan made much the same point as follows:
So the platelet is a cellular entity. It has specific receptors for prostacyclin. When prostacyclin is engaged by those receptors it initiates specific signalling inside the platelet that basically increases levels of factors that help keep the platelet quiescent. So as one individual platelet goes bumping around through your vasculature, through your arteries, through your capillaries, through your veins, it’s exposed to a variety of situations where it might be predisposed to activate but that prostacyclin that’s in the vasculature helps keep the platelets quiet. If you don’t have that prostacyclin, then theoretically they are more likely to aggregate – or activate than aggregate in the presence of any factor that stimulates them whether it be collagen or thromboxane or ADP or any other of a variety of noxious factors that stimulate platelets to become activated.
Both Profs Celermajer and Vaughan proposed that, if someone were in a potentially thrombotic state, and if the source of prostacyclin were turned off, then, if the FitzGerald hypothesis were valid, one would almost inevitably see an elevated production of thromboxane.
501 Returning to the point being made by counsel for the respondents as to the relationship between the administration of a coxib and the production of platelet thromboxane, Prof Celermajer said:
I am sure I see what you are getting at, Mr Garling, which is if there is an imbalance that creates a prothrombotic environment the necessary consequence of a prothrombotic environment is some thrombosis and if thrombosis occurs, platelets will release thromboxane. So you must, if you hypothesise a prothrombotic balance, find thromboxane levels increase because of the thrombus that results from the imbalance, and none of the data that we have seen show urinary thromboxane metabolites going up. So it supports the idea that this is a hypothetical imbalance that actually has no prothrombotic consequence, because if it had a prothrombotic consequence you would have thrombus and you would have thromboxane go up …
Prof Celermajer said “in the McAdam study where they have measured very specifically the platelet thromboxane it is unchanged in the presence of Vioxx”. Prof Celermajer was here referring to the measurement of serum TxB2 rather than to the urinary metabolite (the latter being a measure of systemic thromboxane only). He clarified his point as follows:
I think the paper itself makes this abundantly clear and I will just read two sentences from the abstract … The first sentence is the “Background” section:
We further hypothesised that if the overproduction of prostacyclin in smokers were restraining platelet activation, then inhibition of COX-2 would lead to an increase in the activation of platelets with a corresponding increase in the biosynthesis of thromboxane.
So their hypothesis is if you have an imbalance then removing the imbalance will activate the platelets. The next sentence I wish to read you is from the end of the methods and results:
Levels of serum (TxB2) were not different in smokers and non‑smokers and were unaffected by rofecoxib.
So the hypothesis that was set up in sentence one is unproven by the study which suggests that the hypothesis is incorrect. The hypothesis, … is –
… if the overproduction of prostacyclin in smokers were restraining platelet activation then rofecoxib would increase platelet activation …
… and that is unproven by the study.
Prof Vaughan added, on this issue of the response of thromboxane to the administration of a coxib:
The first author of this publication, Brendan McAdam, was a fellow in my division in cardiovascular medicine in Vanderbilt when this paper was completed, in fact some of the measures performed in this study were done in my own laboratory. We measured the c‑reactive protein and other markers of platelet activation for Brendan. Brendan’s hypothesis was accurately summarised by Professor Celermajer there. They expected to see the presence of an augmented thrombotic state by delivering Vioxx to the individuals in the study and they thought they would [prove] the FitzGerald hypothesis … that prostacyclin would go down and in this higher risk group there would be evidence of platelet activation systemically and that thromboxane levels would go up. In fact, it did nothing and Brendan was astonished that that was the result.
502 Prof Zipes agreed with his colleagues’ exposition of the mechanism of the interaction between the activation of platelets and prostacyclin in the non‑medicated vasculature. He continued:
However, that interaction between the reduced prostacyclin and elevated thromboxane platelet derived, if it produces a thrombosis, it does it locally and that local event is exactly as my learned colleagues have described in terms of activating the platelets locally. This is not a systemic indication that would increase thromboxane levels to a degree that are easily measured but very importantly for the vessel involved, if it happens locally it could still occlude the vessel but you would not get a measurable increase in thromboxane. It’s all in a very tiny area. So I don’t see how or why this should be in conflict with the overall FitzGerald hypothesis.
Prof Harper, who otherwise was not involved in this aspect of the controversy, added:
I just think it’s important to emphasise from a broad perspective if someone is taking Vioxx they are not immediately going to clot things off. The process only becomes relevant when a stimulus for platelet aggregation occurs and if the stimulus is great, platelet aggregation will occur irrespective. If the stimulus is small then perhaps it wouldn’t have occurred to the same extent, but in the presence of Vioxx, because of the mechanisms described, it occurs to a greater extent than it otherwise would.
503 It does seem to have been accepted by Profs Zipes and Harper that, if the FitzGerald hypothesis were sound, one would expect to see an increase in the generation of platelet thromboxane consequent upon the administration of a COX-2 inhibitor. That such an increase has not, apparently, been seen in any human context that might be considered to be presently relevant does, in my opinion, compromise to an extent the utility of the hypothesis to provide a mechanistic basis for the risks associated with the consumption of Vioxx contended for by the applicant. I could not say that the hypothesis itself must be taken to have been disproven – that would be a scientific rather than a legal judgment, and is beyond the court’s role. To the extent that the applicant relies on the hypothesis as part of his legal case, however, I would have to say that the issues discussed in this part of my reasons justify the non‑scientific conclusion that the validity of the hypothesis is a matter of vigorous controversy amongst the experts, and that the rationale relied on by opponents of the hypothesis does seem to be intelligible, coherent and justified by the empirical evidence to which they point.
504 I turn next to the respondents’ seventh point, namely, that the balance between prostacyclin and thromboxane is critical for preventing thrombosis. In their written submissions on this point, the respondents referred to the evidence with which I have dealt above under their sixth point (the effect of coxibs on thromboxane levels). This was not, however, the sense in which the point was put to the cardiologists in concurrent evidence. That was confined to a debate as to the consequences – hypothesised and observed – of the concurrent administration of aspirin with a coxib (item (d) on Prof Celermajer’s list of problems).
505 All the cardiologists provided an affirmative answer to the following question:
I think everyone has agreed that there are multiple systems assisting clotting and haemostasis in the body of which the prostacyclin and thromboxane balance is one but would you agree for FitzGerald as a hypothesis to be scientifically correct that it must be critical for that balance to exist to prevent thrombosis?
The cardiologists were then asked whether, if the balance were critical as proposed, the use of aspirin would be expected to neutralise any adverse effect from the selective inhibition of COX-2. Prof Harper’s answer was to say that there was “evidence that aspirin does not protect against the deleterious effects of COX-2 inhibitors”. Prof Zipes accepted that there was such evidence, but he considered that there were explanations for it, not inconsistent with the FitzGerald hypothesis. Before coming to those explanations, I should refer to the material upon which the respondents relevantly relied.
506 In his evidence, Prof Celermajer had previously (ie previously to counsel for the respondents articulating their seven assumptions) pointed to what he described as “two key pieces of data” to demonstrate that, contrary to what one would expect if the FitzGerald hypothesis were valid, the administration of low‑dose aspirin did not appear to have any effect on the occurrence of cardiovascular adverse events in patients taking a COX-2 inhibitor. The first was the meta‑analysis published as Kearney (2006). Included in the trials for which data were available were 84 placebo-controlled trials that allowed concomitant use of aspirin. The authors stated that they “found no significant heterogeneity of the summary rate ratios for vascular events among aspirin users and non‑users … [and] a similar lack of heterogeneity for myocardial infarction, stroke, and vascular death .…” Profs Zipes and Harper indicated that they were aware of the meta‑analysis by Kearney and others. It was then put to Prof Zipes that, if aspirin were not providing that protection, “that tells against the scientific validity of the FitzGerald hypothesis”. He agreed that it might, but he said that it was “[known] from from well done studies that aspirin may not always protect and there are good explanations as to why it may not work.” He provided four reasons why aspirin might not neutralise the harmful effect of COX-2 inhibition without disproving the FitzGerald hypothesis. They were that (1) the dose of aspirin taken may not have been sufficient in the trials which demonstrated that the administration of aspirin made no difference, (2) there may, in such trials, have been concomitant administration of ibuprofen or naproxen, (3) the coxib itself may biochemically interfere with the beneficial work of aspirin, and (4) to some extent thromboxane may be synthesised by COX-2 in inflammatory cells, additionally to the normal course of synthesis by COX-1 in the platelets (and, implicitly, in such a situation the acetylation of platelet COX-1 by aspirin would not mean the end of thromboxane production).
507 Prof Celermajer’s other piece of data was the study of Nussmeier (2005) in the context of patients who had had CABG. As I have indicated, all patients were taking aspirin, but the relative risk of encountering a cardiovascular event as between continuous coxib use and placebo was 3.7 notwithstanding. In their published article, Nussmeier and colleagues said:
Cardiopulmonary bypass increases the levels of both prostacyclin and thromboxane A2. … However, administration of aspirin, as in our study, theoretically inhibits the formation of thromboxane by platelets. Low‑dose aspirin prevents myocardial infarction and stroke, … and Mangano … has shown that postoperative administration of aspirin is associated with a reduced risk of death and cardiovascular and cerebrovascular ischemic complications after CABG requiring cardiopulmonary bypass. Although our study protocol required the administration of 75 to 325 mg of aspirin daily, resistance of platelets to aspirin is known to occur after CABG. … These aspirin doses, administered concurrently with a selective COX-2 inhibitor after CABG, may have been insufficient to block the formation of thromboxane by platelets in some patients. … Also, 7 of the 20 thromboembolic events (35.0 percent) occurred at least two days after all study medications had been discontinued. Another factor may be thrombocytosis, which is common within two weeks after surgery. … In clinical conditions of enhanced platelet regeneration, the prevalence of COX-2‑dependent synthesis of thromboxane may be increased ....
I have set out this lengthy passage because, as I understood him, Prof Zipes would wish to associate himself with the authors’ explanations of why, in this particular study, the administration of aspirin did not apparently mitigate the adverse impact of the coxib and of why, therefore, the result of the study was not necessarily inconsistent with acceptance of the FitzGerald hypothesis.
508 Of Prof Zipes’ explanations for the apparent lack of impact by aspirin, Prof Celermajer said:
… it seems to me [that] both Professor Zipes and Professor Harper … are using the results of outcome studies to justify an interpretation of mechanistic data, whereas in fact what I think you are trying to ask is if one assumes the mechanistic data are correct, that aspirin inhibits COX-1 and so co‑administration of aspirin removes the imbalance postulated by the FitzGerald hypothesis, then you would expect certain outcomes that weren’t observed in Kearney and Nussmeier, for example. So my view is that the answers that Professors Zipes and Harper have given have used the outcomes to justify the mechanism rather than predicting what the mechanism should show and then examining if the outcomes of the study support that.
Specifically in connection with Prof Zipes’s fourth reason (in para 506 above), Prof Celermajer said:
[T]hat in no way invalidates my argument because in someone on aspirin and Vioxx, COX-1 is gone and COX-2 is gone so prostacyclin is gone and thromboxane is gone, from whatever source. It’s predominantly from platelets and it’s gone because of COX-1 inhibition and a little bit comes from macrophages but that’s gone with COX-2 inhibition. So if you are taking aspirin with Vioxx, the imbalance is gone and if the imbalance is gone and there is no difference in observed events the FitzGerald hypothesis is untenable.
Prof Vaughan stated his opinion as follows:
I think Kearney’s analysis really points to a very important issue. I mean, if anything, you would expect to see a beneficial effect of aspirin in the setting of COX-2 inhibition because theoretically, at least according to the FitzGerald hypothesis, prostacyclin production is reduced, therefore those patients would be exquisitely sensitive or there would be a therapeutic effect of aspirin. The fact that aspirin doesn’t make any difference says again that the hypothesis is non‑operational.
509 Finally on this question, Prof Zipes relied on the following passage in Grosser (2006):
Finally, the issue of a mitigating effect of low‑dose aspirin has not been addressed in the RCTs [randomized controlled trials]. This seems biologically plausible as COX-l knockdown in mice which genetically mimics the impact of low‑dose aspirin … attenuates the prothrombotic and hypertensive effect of COX-2 inhibition .… However, aspirin use was only prespecified in one of the RCTs in humans: TARGET. As mentioned, this was under‑powered to address the cardiovascular question. However, the available evidence is compatible with risk attenuation; the relative risk of myocardial infarction was reduced from 2.37 in nonusers to 1.36 in those patients taking aspirin when lumiracoxib was compared with naproxen. However, this might also reflect a differential capacity of naproxen versus lumiracoxib to interact with and undermine the antiplatelet effect of low‑dose aspirin ….
Counsel for the respondents made the point, which Prof Zipes accepted, that the authors of this article noted that the study referred to (TARGET) was underpowered in relevant respects. Notwithstanding that, Prof Zipes contended that there were indications, contrary to what appeared from the meta‑analysis by Kearney and others, that low‑dose aspirin might be mitigatory.
510 The respondents invited me to find that the FitzGerald hypothesis could not be sound for reasons which involved the demonstrated absence of mitigation by the administration of low‑dose aspirin. However, that is itself an issue upon which I could not find that there was a scientific consensus. It is not just that Prof Zipes resisted the conclusion drawn by Profs Celermajer and Vaughan, it is also that he derived support in that resistance from the observations (albeit guarded and qualified) of the authors of the Nussmeier and Grosser papers. Notwithstanding that, there appear to be quite solid empirical data (ie those referred to by Kearney and colleagues) that are inconsistent with the hypothesis that coxibs are pro‑thrombotic because they leave thromboxane unaffected while suppressing prostacyclin. In this state of scientific uncertainty, I take the view that the absence of demonstrated mitigatory impact of low‑dose aspirin stands in the way of an unqualified acceptance of the FitzGerald hypothesis as a mechanistic explanation for the empirical data on which the applicant relies.
511 The next of the respondents’ points to which I propose to turn is that the FitzGerald hypothesis required that a partial reduction in prostacyclin would have a clinical effect. Each of the cardiologists agreed with that as a proposition. Counsel for the respondents then put the following question to Prof Zipes:
Isn’t it the fact that even a 90 per cent decrease of prostacyclin released by endothelial cells still leaves sufficient prostacyclin to prevent platelet aggregations in humans?
It was put to Prof Zipes that, if that statement (which corresponds with item (e) on Prof Celermajer’s list of problems) were correct, it would stand “as a large demonstrative barrier to a successful demonstration of the FitzGerald hypothesis”. Prof Zipes did not agree. He said: “It’s going to depend upon where the prostacyclin is as to its interaction with thromboxane”. When asked if he supported the proposition that a 90% decrease of prostacyclin still left enough prostacyclin to prevent platelet aggregation in humans, Prof Celermajer said: “In the circumstance where that study was performed, yes”. Prof Vaughan agreed.
512 The study to which Prof Celermajer referred was that published as Jaffe (1979). Of that study, Prof Celermajer said:
Jaffe and Weksler … noted that even a 90% decrease of prostacyclin release by endothelial cells still left enough prostacyclin to prevent platelet aggregation in humans in vivo. Thus one would have to postulate a very profound inhibition of prostacyclin indeed, and establish that no compensatory mechanisms through other enzyme systems such as [nitric oxide] synthase were invoked, for the FitzGerald hypothesis to hold true.
Prof Zipes’ response to this statement was as follows:
What [Prof Celermajer] does not include is that this statement, taken from a 30‑year old study, was based on projected (not measured) PGI2 levels calculated from the assumed surface:volume ratio of the normal human vascular system. Jaffe and Weksler published no experiments in this study to prove that statement. Further, the calculations were computed from estimates in a theoretical normal individual and, therefore, with normal TXA2 levels. Obviously, if TXA2 levels were elevated in an atherosclerotic individual, the assumptions would not hold.
513 Exactly what Prof Celermajer meant by “the circumstance where that study was performed” was not clarified. Some light is thrown on the subject by a consideration of the (previous) evidence of Prof Cleland given in the rheumatology concurrent session. The following passage from the Jaffe article was referred to by counsel for the respondents:
Thus in vivo production of PGI2 at 10% of maximum could theoretically yield a peak blood PGI2 concentration of [about] 830 nM (2.8 nM x 2.86 x 103/9.6). In vitro, such levels of PGI2have a profound inhibitory effect on platelet behavior. For example, 1.4 nM PGI2 fully inhibited thrombus formation, and 57 nM PGI2 inhibited platelet adhesion by 56% when human blood was exposed to rabbit aorta subendothelium [reference given]. Furthermore, 47 nM PGI2 caused a half‑maximal increase in platelet cyclic AMP to a level of [about] 12 times basal level [reference given] when only a doubling of cyclic AMP was needed to inhibit platelet aggregation [reference given]. Similarly, we have reported that 0.1 nM PGI2 completely inhibited platelet aggregation in platelet‑rich plasma induced by threshold levels of arachidonic acid [reference given]. Thus, in man and experimental animals even a small fraction of the productive capacity for PGI2 in normal blood vessels should result in more than enough PGI2 to inhibit platelet function.
Prof Cleland responded by pointing out that, in their comment about the ability of rather low levels of endothelial prostacyclin to inhibit platelet aggregation, the authors were referring to studies other than that as to which the article was directly written. He said that it would be more informative to look at those other studies. He continued:
The issue about in vitro studies is they help you to explore mechanism and show you what’s possible. They don’t allow you to predict accurately what happens in the whole body. They certainly give you a guide as to what to look for and what might happen but there are a lot of uncertainties here. Here we have early methods based on endothelial cell culture in artificial culture media. There’s no issues of COX selectivity addressed here.
Pressed to respond to the conclusion by Jaffe and Weksler that “the ability of endothelial cells in normal blood vessels to produce PGI2 seems to be far in excess of that needed to inhibit platelet function”, Prof Cleland said:
I don’t think that’s necessarily correct. These studies were undertaken in the era when they were trying to sort out optimal doses for low dose aspirin and really, what they are arguing is even though aspirin has some systemic effect on prostacyclin production it’s not sufficient to override a possible beneficial effect of taking low dose aspirin on the platelet. The issue as to whether sustained long term inhibition of systemic prostacyclin production via COX-2 has adverse cardiovascular effects in a minority of patients, a minority to those affected, isn’t excluded by these studies.
Profs Celermajer and Vaughan were not taken to this evidence, and said nothing more about the Jaffe article than I have indicated above.
514 I must say that the respondents’ final written submissions dealt with this point as to the clinical effect of a partial reduction in prostacyclin in a way that, to put it most charitably, did not reflect the thrust of the questions put to the cardiologists or to Prof Cleland. Those submissions referred to the evidence given by Profs Celermajer, Vaughan and Zipes, but contained no argument as to why I should accept the proposition implicit in the respondents’ reliance on the Jaffe article. The submissions merely asserted, without elaboration, that Prof Zipes had “relie[d] on weak criticisms to argue that the study is flawed”. I did not understand Prof Zipes so to contend.
515 The respondents’ submissions proposed that their experts had “concluded that a vascular prostacyclin reduction of greater than 90% would be required to prevent platelet aggregation”. As I understood the position with which Profs Celermajer and Vaughan implicitly agreed in concurrent evidence, however, it was that a vascular prostacyclin reduction of greater than 90% would be required to permit platelet aggregation. The submissions then argued that Prof Celermajer had “put it most succinctly” when he said:
It’s fanciful, in my view, to suggest that ... the relatively modest inhibition of prostacyclin production by Vioxx – which even if you believe Catella‑Lawson is only around 60 per cent – would sway the balance.
The submissions relied also on Prof Vaughan: “… to think prostacyclin has to do all that work on its own against an infinite excess of thromboxane is really almost – it’s unimaginable”; and “… if you inhibit prostacyclin production it doesn’t really matter because there’s so much thromboxane there in the first place. Even if you didn’t inhibit it, it can’t really counteract what’s happening specifically at the site of a clot …”.
516 Thus, as it seems to me, the respondents are simultaneously tying themselves to two contradictory positions in proposing that a partial reduction in prostacyclin will not have a clinical effect. Ironically perhaps, either position, if valid, may well justify such a proposition. The first position (the one put to the cardiologists in concurrent evidence) is that even a reduction of 90% would not have a clinical effect in the sense that the remaining 10% would be sufficient to inactivate platelets. The second position (that most clearly relied on in the respondents’ written submissions) is that, even in the absence of any reduction, endothelial prostacyclin is unlikely to be sufficient to cope with what Prof Vaughan described as “an infinite excess of thromboxane”. That position appears to be consistent with Prof Harper’s evidence that there is a distinction between great and small stimuli to platelet aggregation. As he said (see para 502 above) if the stimulus is great “platelet aggregation will occur irrespective”. This is, I consider, an important point, and I shall return to it. For present purposes, however, my conclusion is that the mechanism proposed by the FitzGerald hypothesis, if otherwise consistent with the science, is likely, at least in some circumstances, to have a clinical effect.
517 I turn finally to the first of the respondents’ points, namely, that the FitzGerald hypothesis implies that compensatory systems in the vasculature are insufficient to overcome the prothrombotic effect caused by an imbalance between prostacyclin and thromboxane (item (i) on Prof Celermajer’s list of problems). Prof Celermajer’s position was effectively that upon which this point was based, and was set out in his report:
In addition to the prostacyclin/thromboxane system, the classical coagulation pathway, platelets, the [nitric oxide] system and the plasminogen activation/activation inhibition systems are all relevant components to a delicately balanced homeostasis for humans, which has the vital function of preventing excessive bleeding and excessive thrombosis. It is unlikely that perturbation of any single system would not result in some counter‑balancing compensatory change of another system and there are extensive data to suggest a degree of cross talk between several of these systems, especially the prostaglandin and [nitric oxide] systems.
Prof Vaughan’s position was as follows:
It’s rather interesting to look at important clinical trials that have examined the relative relationship between arterial or vasodilator capacity – arterial vasodilator capacity which is mediated by nitric oxide. For example, if you have an impairment in flow mediated vasodilation that is a predictor of cardiovascular events over time. The nitric oxide system is one of the systems that helps protect against thrombosis. So not only does it plausibly impact upon things like platelet aggregation, there’s clinical data to indicate that impairments in flow mediated vasodilation project risk. … There is another system that contributes to our ability to protect ourselves against intervascular thrombosis, that’s the plasminogen activator system. It involves the release of [tissue plasminogen activator] from our endothelial cells which facilitates the dissolution of clots. Impairments in arterial [tissue plasminogen activator] release are associated with increased cardiovascular events over time.
As the respondents pointed out in their written submissions, Prof Vaughan resisted the proposition that the different vascular systems here under discussion were, as proposed by Prof Celermajer, “delicately balanced”.
518 Profs Zipes and Harper accepted that the regulation of thrombosis and bleeding involved a complex interaction in the human system and that “in the classical coagulation pathway there [were] a number of relevant components” including the platelet system, the nitric oxide system, the plasminogen activation systems and the activation inhibition systems. They accepted that that each of those pathways had extensive “cross‑talking or cross‑connection between them with one system leading to compensatory changes in the others” and that there were “multiple lines of defence to clotting in an artery”. However, Prof Zipes did not accept the next proposition put to him by counsel for the respondents, namely, that “the FitzGerald hypothesis assumes that by altering but one of the multiple systems for clotting the others will not undertake any compensatory effect”. He said that “the FitzGerald hypothesis could clearly be operative and overpower whatever compensatory mechanisms are there or interact with those mechanisms in some fashion”. When asked if he knew of any study that established that the compensatory mechanisms were insufficient to overcome the imbalance proposed in the FitzGerald hypothesis, Prof Zipes said that “when you have a myocardial infarction you have completed the essential experiment and that imbalance has led to a thrombus that [oc]cluded a vessel”.
519 Prof Harper’s response was more lengthy, and, because of its importance, should be set out in full. He said:
In any system there is always [a] compensatory system but if an outcome occurs such as a thrombus then clearly the compensatory system hasn’t been sufficient. I think in the setting that we are describing a plaque rupture is a very strong stimulus to clot formation and therefore you need – the compensatory system has to be strong to overcome it. But the degree of stimulus depends on – not all plaque ruptures are the same so in some plaque ruptures the stimulus is so strong because of exposure of collagen, et cetera, that nothing can overcome it, and irrespective of whether a patient is or isn’t on Vioxx some plaque ruptures will clearly lead to a heart attack. But it’s possible that some plaques are ruptures that would otherwise not lead to an event. If the stimulus for thrombosis is slightly more, is slightly greater, then in that particular case the thrombus will occur, and my concept is that Vioxx does increase the risk of thrombosis. I am not convinced that it’s necessarily the FitzGerald hypothesis, and that that sometimes leads to an event that otherwise wouldn’t occur.
The sense of what Prof Harper most recently said in this passage would have been more accurately rendered in transcript as “… my concept is that Vioxx does increase the risk of thrombosis (I am not convinced that it’s necessarily the FitzGerald hypothesis) and that that sometimes leads to an event that otherwise wouldn’t occur.”
520 There was no resolution of the disagreement amongst the cardiologists on this point, most probably because it effectively lies at the core of the FitzGerald hypothesis itself. That is to say, whereas Profs Zipes and Harper, following FitzGerald, hypothesised that a disturbance of the balance between endothelial prostacyclin and platelet thromboxane might well lead to thrombotic consequences, Profs Celermajer and Vaughan proposed a number of reasons why it need not. Profs Zipes and Harper accepted the existence of the compensatory systems to which the others referred, but adhered to their position that the inhibition of COX-2 was nonetheless a factor – albeit one amongst several – that would have an impact in a certain direction. They were never likely to concede that the existence of the other systems to which Profs Celermajer and Vaughan referred would eliminate the potential for that impact, and they did not.
521 Overall, the burden of the evidence of Profs Celermajer and Vaughan was that the FitzGerald hypothesis was, as an explanation of the consequences of the inhibition of COX-2 in the vasculature, still no more than a hypothesis. It had not been scientifically validated. Profs Zipes and Harper accepted that, but contended that, as such, it was a biologically plausible explanation for what was observed in APPROVe and elsewhere. Prof Harper’s position was that the data yielded by APPROVe established that the consumption of Vioxx increased the risk of CVT events, and that it did not matter greatly whether Dr FitzGerald’s hypothesis was the explanation. He did, however, consider the hypothesis to be plausible. Prof Zipes’ position was somewhat more categorical, in that he regarded the hypothesis as the only plausible explanation for the data currently known to science and, in the absence of a better explanation, he considered that the hypothesis should be accepted.
522 In assessing the plausibility of the FitzGerald hypothesis as an explanation for what was observed in APPROVe, one must, in my view, keep in mind that we are here concerned to identify an explanation for adverse events which occurred in a small minority of the patients who took Vioxx. Even taking account of the extended range of data looked at in Baron (2008), of the 1287 patients in the Vioxx arm, only 59 suffered CVT events (of which 34 were myocardial infarctions) in the period under review. On any view, therefore, it could not be said that taking Vioxx “caused” these endpoints in the sense of giving rise to them as a direct and inevitable consequence. That is not a complete answer to the applicant’s case, of course, but it provides a context for assessing questions of plausibility. It is not surprising that, under certain conditions and in certain study settings, outcomes which might reasonably be expected if the FitzGerald hypothesis were valid are not consistently observed.
523 Summarising the position reached after consideration of the respondents’ seven points which were said to confound the FitzGerald hypothesis, I would commence by noting that there is no scientific consensus as to the presence of COX-2 in the endothelium or as to its activity in a situation of atherosclerosis. As noted earlier in these reasons, prostacyclin acts only in the immediate vicinity of its synthesis, which means that, if it were to have an effective anti‑aggregatory role in the platelets, it would need to be synthesised in the endothelium at, or closely adjacent to, the point of interest. It seems that, scientifically, whether this is so remains a matter of hypothesis.
524 One piece of pharmacological evidence that would at least have pointed to the validity of the FitzGerald hypothesis would, it seems, have been an increase in the production of platelet thromboxane in response to the inhibition of COX-2 in the context of an inflamed vasculature. However, the researchers who studied this aspect found no such consequence. Profs Zipes and Harper pointed out that that was not conclusive, a proposition which I accept. But the result does add to, rather than reduce, the uncertainty associated with the consequences in the vasculature of inhibiting COX-2 while sparing COX-1.
525 The mitigation of the hypothesised pro‑aggregatory effect of selectively inhibiting COX-2 by the administration of low‑dose aspirin would be another mark of consistency between the FitzGerald hypothesis and the empirical experience. No such mitigation, however, has been observed. Again, there may be reasons for this which do not involve the rejection of the hypothesis, but neither can we mark down the aspirin effect as a sign of consistency of the hypothesis with actual experience.
526 That brings me to the final two points raised by the respondents as to the FitzGerald hypothesis. They concern the linkages between the mechanism proposed by the hypothesis and the clinical outcomes potentially produced. Here there are, it seems, two aspects to consider. The first relates to the characteristics of the event or events in the vasculature which give rise to the presumptive thrombus. The second relates to the system’s biological response to those events. Both are, I consider, important to an assessment of the hypothesis generally and to the attribution of a causal relationship between COX-2 inhibition and a CVT event in a particular case.
527 As to the first aspect, I refer to the evidence of Prof Harper which I have set out at paras 502 and 519 above. Of all the cardiologists, Prof Harper had, if I may observe without disrespect to his colleagues, perhaps the most conspicuous focus upon clinical, as distinct from experimental, circumstances. He made it clear that, whether or not the FitzGerald hypothesis is valid, we are not here talking about absolutes. In the unmedicated vasculature, the stimulus to platelet aggregation following plaque rupture may be weak, or it may be strong. A small or large thrombus may result. It may lead to an occlusion of an artery more or less immediately, or it may do so later, possibly in conjunction with other prothrombotic circumstances then existing. However these factors may work out in the normal case, when the selective inhibition of COX-2 is added to the mix, in Prof Harper’s view there is now another pro‑aggregatory factor which, in all cases, makes it that much more likely that what would not otherwise have been a CVT event becomes one and that a CVT event that would in any event have occurred occurs sooner.
528 As to the second aspect, all the cardiologists were agreed that the vasculature has a number of systems which tend to protect it against the consequences of platelet aggregation. If the FitzGerald hypothesis is valid, the up‑regulation of endothelial COX-2, resulting in the production of prostacyclin, is one of them. In that event, the selective inhibition of COX-2 would leave the vasculature with one less defensive mechanism than it has in the unmedicated state. That was the position for which Profs Zipes and Harper (and, indeed Prof Cleland) contended. But the continued existence (and importance) of the other defensive mechanisms – together with the heterogeneous nature of aggregatory stimuli –means that the inhibition of COX-2 will not necessarily render thrombotic all events or circumstances which otherwise would not have been so. If the FitzGerald hypothesis is valid, it may be supposed that, over a population of patients, there will be some for whom the inhibition of COX-2 will make a difference, but it seems that there may not be a way of knowing before the event who those patients will be or of diagnosing after the event who those patients were.
529 Starting from the position that, if the FitzGerald hypothesis is valid, it makes a statement about the course of thrombus formation after plaque rupture, the position, therefore, seems to be more or less as follows. In the unmedicated vasculature, some ruptures generate strong stimuli which lead to thrombi, even with prostacyclin doing its presumptively compensating work. In the context of selective COX-2 inhibition, some ruptures generate weak stimuli which do not have thrombotic outcomes because of the compensatory work of the other systems to which Profs Celermajer and Vaughan referred, notwithstanding the absence of prostacyclin. Between these extremes, it is postulated that there are some stimuli which are of a strength, and which occur in cardiovascular settings generally, as would be effectively counteracted by the compensatory systems as they exist in the unmedicated vasculature, but where the removal of prostacyclin would make a clinical difference. If I am correct in supposing that there are these various combinations and permutations, it is not surprising that the studies that have looked at the cardiovascular consequences of COX-2 inhibition have produced varying results.
530 Does the FitzGerald hypothesis provide a plausible biological explanation for the excess of CVT events observed in the results of APPROVe and the other studies to which I have referred? Before answering that question, I propose to consider the remainder of the applicant’s submissions on the biological issues, as a consideration of them has the potential to throw light on the relationship between the results of the trials and studies in which Vioxx was involved and the mechanism on which the applicant relies.
Other aspects of a biological explanation
531 The applicant submitted that Vioxx “contributed” to each of the four stages of atherosclerosis identified by Prof Zipes. In this respect, he relied first upon the following statement by Dr FitzGerald himself in what Prof Zipes called an “opinion piece” written in 2004:
Thus, a single mechanism, depression of prostaglandin I2 formation, might be expected to elevate blood pressure, accelerate atherogenesis, and predispose patients receiving coxibs to an exaggerated thrombotic response to the rupture of an atherosclerotic plaque.
A reading of Dr FitzGerald’s piece, however, shows that he used the expression “might be expected to …” as an indication of what his research group hypothesised in 1999. Be that as it may, the passage above refers to three respects in which Vioxx might have harmful effects: the elevation of blood pressure, the acceleration of atherogenesis and the exaggeration of the thrombotic response to plaque rupture. The third is the concern of the FitzGerald hypothesis itself. The other two were also the subject of direct evidence by the cardiologists in this case.
532 The cardiologists were asked for their opinion on the question whether Vioxx affected blood pressure. In their joint report, they said that Vioxx did increase blood pressure in some subjects, adding that they did not believe –
… that the blood pressure raising effect of Vioxx is significantly different from that of other NSAIDs. As these drugs affect salt and water handling by the kidney, their effects on [blood pressure] are broadly similar.
Profs Zipes and Harper, however, added that they considered that the effect of Vioxx on blood pressure was greater than that of Celebrex, and in another part of the joint report they expressed the view that Vioxx had a more pronounced hypertensive effect than either Celebrex or other NSAIDs. Profs Celermajer and Vaughan disagreed, adhering to the view that Vioxx did not cause more hypertension than other NSAIDs.
533 For his view that Vioxx contributed to hypertension, Prof Zipes relied on a meta‑analysis of 19 RCTs involving coxibs published as Aw (2005). The relative risk of developing hypertension from taking Vioxx rather than placebo was found to be 2.63 (1.42, 4.85) and the corresponding figure where the comparator was the non‑selective NSAIDs combined was 1.78 (1.17, 2.69). In his statement in reply, Prof Vaughan said that the Aw study contained “several methodological limitations”, most importantly the inclusion of RCTs in which Vioxx had been taken at doses higher than any that would be recommended to be used chronically. When limited to studies that involved Vioxx at 25 mg per day, the picture that emerged was of “a modest association with increases in blood pressure” only. In their final submissions, counsel for the applicant made no reference to this evidence by Prof Vaughan, and he was not cross‑examined on it.
534 In her statement in reply, Dr Reicin addressed Prof Zipes’ conclusion that Vioxx increased blood pressure in some detail. She said that it had long been well‑known that NSAIDs increased blood pressure in some patients, and that the effect was dose‑related. Even when dose was taken into account, NSAIDs appeared to have a range of blood pressure effects, with naproxen “at one end of the spectrum”, ibuprofen at the other end and diclofenac “somewhere in the middle”. (Dr Reicin did not say which end was which.) Dr Reicin referred to data contained in Merck’s Integrated Summary of Safety that accompanied its New Drug Application to the FDA in November 1998. According to Dr Reicin, those data –
… showed that the incidence of hypertension‑related adverse experiences in patients taking 25 mg Vioxx was comparable to the incidence seen in patients taking other NSAIDs, and not unexpectedly higher than the incidence seen in patients taking placebo.…
….
The data also showed that Vioxx use was associated with small increases of systolic blood pressure…[of] the magnitude of <3 mm Hg … on a population basis, and “[t]here was no clinically important difference between … [Vioxx] and comparator NSAlDs in their effect on blood pressure.”
Dr Reicin referred also to the Background Package prepared by Merck for the 2005 meeting of the FDA Advisory Committee. In that material, Merck had said:
Clinical trials with rofecoxib at daily doses of 12.5 and 25 mg in patients with osteoarthritis have shown effects on hypertension and edema similar to those observed with comparator NSAIDs. … In clinical trials of rofecoxib at daily doses of 25 mg in patients with rheumatoid arthritis the incidence of hypertension was twice as high in patients treated with rofecoxib as compared to patients treated with naproxen 1000 mg daily. A difference from naproxen was not reproduced in the ADVANTAGE study in OA patients. … Similar to clinical doses of other NSAIDs, use of rofecoxib 25 mg is associated with small increases compared to placebo in mean systolic blood pressure <5mm Hg and mean increases of mean diastolic blood pressure <2mm Hg.
Dr Reicin also mentioned the hypertension data in the VIGOR trial, where a higher incidence of hypertension among Vioxx patients than among the naproxen patients was observed. She added that this was not surprising, since NSAID‑related blood pressure increases were generally dose‑dependent, and the 50 mg Vioxx dose in VIGOR was twice the recommended chronic dose. Dr Reicin was not cross‑examined on any of this evidence.
535 Absent a challenge to the evidence of Prof Vaughan and Dr Reicin to which I have referred, I do not think I am in any position to reject that evidence. I could not make a finding any more favourable to the applicant than that Vioxx did contribute to hypertension in some people, but not to an extent that would put it outside the range of contribution by NSAIDs generally.
536 The cardiologists were asked for their opinion on the question whether Vioxx accelerated atherosclerosis. They commenced by noting that Vioxx caused hypertension in some individuals, and hypertension was known to accelerate atherosclerosis. They added: “The same argument applies to all selective or non‑selective inhibitors of the COX system.” Save for this aspect, the cardiologists agreed as follows:
There are no studies in humans that have directly measured the progression of atherosclerosis in coronary arteries following Vioxx administration. The available animal data with regard to COX-2 inhibition are conflicting with some studies showing progression and others retardation of atherosclerosis.
Profs Celermajer and Vaughan said that the (animal) studies involving Vioxx of which they were aware showed that the drug either had no effect, or had an effect by way of reduction, on the development of plaque.
537 Prof Zipes went further than the other cardiologists in identifying the range of mechanisms by which the inhibition of endothelial COX-2 might be expected to accelerate the development of atherosclerosis. The first was that it accelerated endothelial injury. In his report, Prof Zipes said that a “major source of endothelial injury is increased shear stress due to hypertension.” He considered the blood pressure data from the APPROVe study, and concluded: “Through its promotion of hypertension, Vioxx contributes to the progression of atherosclerosis both in its earliest (endothelial injury) and latest stages (complicated atheroma)”. In a summary of the effects of Vioxx, Prof Zipes said that “endothelial injury is accelerated through increased exposure to hypertension”. Save for a reference to a study of mice to which I shall return, the only basis upon which Prof Zipes’ report proposed that Vioxx caused or accelerated endothelial injury was by way of increased blood pressure. As I have said, that was a mechanism which Vioxx had in common with non-selective NSAIDs.
538 Prof Zipes’ next point related to the formation of the fatty streak. He said that this depended on the movement of LDL cholesterol into the vessel wall. If it were oxidized, it then would be ingested by white blood cells to form foam cells. He referred to Walter (2004), in which it had been found that Vioxx increased the rate and extent of oxidation of human LDL through a non‑enzymatic means. However, in his statement in reply Prof Vaughan pointed out that the study by Walter and colleagues was an in vitro one (a fact accepted by Prof Zipes in his oral evidence) and that the work published as Lekakis (2006), which was a study of Vioxx in vivo in humans, showed a decrease in oxidation. In his oral evidence, Prof Zipes said that he was not familiar with the article by Lekakis et al, and was unable to comment on Prof Vaughan’s evidence. Prof Vaughan concluded this aspect of his statement in reply as follows:
Further, studies investigating the effect of Vioxx on chemicals called isoprostanes that are markers for systemic oxidative stress showed no effect … Thus, although [the] applicant’s experts suggest that Vioxx is detrimental in oxidizing LDL, studies in living humans taking Vioxx simply do not support that assertion.
539 Prof Zipes’ third point was that Vioxx acted to accelerate atherosclerosis because it interfered with the formation of, and thereby led to less stable, fibrous plaque. Whether the destabilisation of atherosclerotic plaque was a physiological mechanism by which Vioxx increased the risk of encountering a pleaded cardiovascular condition was one of the questions asked of the cardiologists. They all said that they knew of no direct evidence in humans which indicated that Vioxx destabilized atherosclerotic plaque. This was confirmed in the concurrent session, but Prof Zipes relied on animal studies in support of his opinion that it did so. One of those was published as Egan (2005) in which (to use Prof Zipes’ words) the authors –
… studied a model of intense atherogenesis, combining apobec‑1 deficient and LDL receptor deficient mice to mimic human atherosclerosis. In this model, antagonism of the thromboxane receptor significantly retarded the development of atherosclerotic lesions, but when combined with a congener of Vioxx, significant evidence of plaque destabilization was seen, a critical step in the progression to plaque ulceration, thrombosis, and infarction.
540 In the concurrent session, Prof Vaughan commented on Egan as follows:
I would like to preface my remarks regarding this publication by saying that first, mice are not necessarily a great replica of human disease. Mice generally don’t rupture plaques and they generally don’t have myocardial infarctions in the setting of atherosclerosis. ... This study by Egan and colleagues recorded a [morphological] difference in the roof of the plaque in mice that were treated with a COX-2 inhibitor, not Vioxx but MF tricyclic, together with a thromboxane receptor antagonist. Those mice apparently had plaques that looked like they might be more susceptible to breakage. The administration of the COX-2 inhibitor alone did not induce those morphological changes so it was a very distinct model and it involved the administration of two drugs and it was only seen when the thromboxane receptor antagonist was given together with the COX-2 inhibitor.
In response to Prof Vaughan, Prof Zipes said that there were other studies (to like effect) to which he could have referred, but refrained from doing so to save time. He agreed, however, that the picture which emerged from the animal studies was confusing “because they go in both directions”. He added: “You can pick a study to support your position”.
541 The other indirect evidence upon which Prof Zipes relied involved what the respondents described as reverse engineering. He said:
But I come back to the outcomes data because without an unstable plaque you don’t have the plaque rupture, you don’t have the thrombus, you don’t have the myocardial infarction so again, reasoning backward from the multiple clinical trials showing an increase of myocardial infarction from Vioxx, it has to be working in some fashion via the atherosclerotic – in a plaque and subsequent thrombus.
I consider, however, that this approach was correctly answered by Prof Celermajer as follows:
Just by way of comment, that you take an eventual putative outcome and go backwards to say that each mechanism that might contribute to that outcome is invoked, if there is a three step process and you are looking at the outcome of the three step process I am not sure why you reason that all three of those steps must be accelerated to get there faster. … If you are running three miles and one day you get there in eight minutes and the next day you get there in seven minutes it could either be because you ran the first mile faster or the second mile faster or the third mile faster. It seems to me that Professor Zipes is imputing all three of the mechanisms – prothrombosis, proatherosclerosis and proplaque rupture – by measuring the one single end point ….
I am not persuaded that the evidence is sufficient to justify a conclusion that Vioxx contributed to the acceleration of atherosclerosis in any or all of the three respects identified by Prof Zipes.
542 However, there was another potential mechanistic link between the action of Vioxx and the acceleration of athersclerosis which was debated in the joint cardiology session. The nature of that link is best illuminated by referring to the evidence of Prof Harper. He agreed that there was no evidence that Vioxx caused atherosclerosis, but considered that the pro‑thrombotic work of Vioxx most probably contributed to the thickening of plaque by a series of minor, clinically silent, events which, over time, accelerated the process of atherosclerosis. He said:
I think it’s analogous to smoking. Smoking is a clear risk factor for heart disease. If you stop smoking the risk doesn’t diminish instantly, it diminishes gradually over time, at least a few years, but it does diminish and I think the situation would – we don’t have the data but the situation would likely be the same with Vioxx. Atherosclerosis is a progressive disorder so over a few years most people will have progress in plaque or their plaques will increase in size over a period of time. I believe that Vioxx probably accelerates that because it makes it more likely that thrombus gets incorporated into the plaque so when you stop Vioxx you have had – you stop at a point when you have more atherosclerosis than you would otherwise have and it will take a while for that effect to wear off.
As I understand his position, Prof Zipes would wish to be associated with these views of Prof Harper. Asked what the FitzGerald hypothesis would say about the circumstances of a patient who, say, took Vioxx for a month and then omitted to take it for the next three months, Prof Zipes said:
Mechanistically [the hypothesis] would then lend credence to the fact that Vioxx played a role in the development of atherosclerosis, so that even stopping it when presumably the prostacyclin levels would come back to normal, you would have the balance and so on. If there were residual actual structural changes in the development of atherosclerotic diseases they would not regress and therefore there would be some residual risk.
Prof Celermajer did not disagree with Profs Harper and Zipes insofar as their evidence dealt with the rate of pace of atherosclerosis, but considered that that issue was unrelated to the mechanism proposed in the FitzGerald hypothesis.
543 Prof Vaughan approached the matter from an experimental, rather than from a conceptual, standpoint. He said:
I was chief of cardiovascular medicine at Vanderbilt for 10 years before I moved to Chicago last year and the three really pivotal studies, experimental studies that looked at the effect of Vioxx in mouse models of atherosclerosis were done in my cardiology unit by Mike Burleigh, I served on his thesis committee, and by my colleague Craig Lynton. They clearly showed – and I have seen the primary data. They showed that Vioxx reduced the development of atherosclerosis in those mouse models very, very consistently and I think those studies will stand out compared to all the other models and all the other experiments relating or describing the role of COX-2 in atherosclerosis. They clearly reduced it, they clearly prevented it and it was very consistent and those mouse models, although not perfectly analogous to human disease, are reasonable representations of what goes on in the vasculature of human beings.
Later in his evidence, Prof Vaughan said:
There are numerous animal studies that have been done, mostly using genetically modified mice to look at the impact and the ramifications of modulation or inhibition of cyclooxygenases on both COX-1 and COX-2 for the development or progression of atherosclerosis. There are five different studies that I am aware of that have looked specifically at Vioxx in these models. All of them show either a reduction in the development of atherosclerosis or no effect. None of them show an increase in the development of atherosclerosis. There are other studies, to be honest, that demonstrate that other drugs that impact on COX-2 activity or other genetic modifications in mice can lead to atherosclerosis but those do not involve the use of Vioxx.
Prof Zipes agreed with that statement.
544 The position, then, seems to be this. There was no real dispute that thrombotic material in the vasculature might well, over time, become incorporated into plaques and thus accelerate the development of atherosclerosis. Although Prof Harper did not suggest that Vioxx had anything to do with that process of incorporation as such, if Vioxx had earlier (ie through the mechanism proposed by the FitzGerald hypothesis) promoted the formation of thrombotic material, it might be thought to have an indirect effect on the acceleration of atherosclerosis. These propositions, however, seem to be conceptual, or logical, rather than empirical. There appears to be no data from studies involving humans which would support them. To the extent that they might be useful, the animal stidies are at best equivocal, and, as Prof Vaughan pointed out, contradictory in situations in which Vioxx itself was used. These uncertainties are serious enough of themselves. But the situation facing the court is made the more problematic by the circumstance that we are here dealing with goings-on in the vasculature at least one remove from the action of Vioxx itself, as proposed by Dr FitzGerald. In the circumstances, I consider that it would be to overreach the science to conclude that Vioxx accelerated atherosclerosis, or to infer that the results of the studies and trials of Vioxx to which I have referred were given a plausible biological explanation not merely by the FitzGerald hypothesis itself but also by a state of affairs in the vasculature which was assumed, conceptually as it were, the more readily to arise if that hypothesis were valid.
545 It is convenient here to deal with a further question which was addressed to the cardiologists, and which they answered, up to a point, in their joint report. They were asked to comment on the manner in which the dose, duration and pattern of use, and patient population, would affect (if at all) the physiological mechanism whereby the consumption of Vioxx increased any such risk of cardiovascular events as they identified. Elsewhere in these reasons, I consider the matters of dose and patient population. I comment on matters of duration and pattern of use here.
546 As to duration of use, Profs Zipes and Harper believed that the adverse effects of Vioxx started early after initiation of therapy, at least within the first month. They considered that the initial interpretation of the time-rate of occurrence of CVT events in the APPROVe study (see para 331 above) had been shown to be incorrect, and that a correct analysis of the data would suggest that the adverse effect of Vioxx began early. They pointed out that, in the VIGOR study, the adverse effects of Vioxx occurred early after the initiation of therapy, and that the results from the meta‑analysis of Juni (2004) gave the same indication.
547 Profs Celermajer and Vaughan said that the data on duration were inconclusive, but noted “the inconsistent patterns of cardiovascular event accumulation in various trials”. Notwithstanding that caution, Prof Celermajer made it clear − and I did not understand Prof Vaughan to take a different position − that, if Vioxx were pro-thrombotic, one would expect that tendency to emerge soon after the commencement of administration of the drug. He said: “Coronary thrombosis is an acute sudden event, it does not develop over 18 months, it does not develop over 18 days”. Although Prof Celermajer expressed this view in the context of a position (which he took) that Vioxx was not pro-thrombotic, to the extent that the FitzGerald hypothesis is considered to provide a plausible biological explanation for the cardiovascular results of the studies referred to above, I think that he and Prof Vaughan would agree with their colleagues that the effect of the drug would be felt soon after the patient concerned commenced to take Vioxx.
548 The matter of pattern of use is more problematic. Here I am concerned, for example, with the case of a patient whose consumption of Vioxx is intermittent, or who takes Vioxx for a time and then discontinues. In their joint report, the cardiologists noted that the cardiovascular effects of the discontinuous use of Vioxx had not been reported in any trial. Indeed, subject to such reservations as were proper to be made about compliance, the assumption upon which the data from the various studies to which I have referred were reported was that Vioxx was consumed as intended: once daily.
549 This aspect was explored further in the cardiology concurrent session. Prof Zipes referred to the follow-up data from the APPROVe trial, which was the subject of Baron (2008) − see paras 338‑339 above − and to the position apparently taken by Dr FitzGerald:
As a matter of fact, that was raised by FitzGerald early on and recommending the continued follow-up of the APPROVe data which was supposed to be intention to treat observations and he made the point with which I subscribe or agree, that if Vioxx had caused some of the changes that we are talking about so that there was a progression of atherosclerosis, that even stopping Vioxx at that point, assuming the individual took the drug long enough, that there would be residual effect and hence, he wanted the follow-up data from APPROVe which I think suggests that he was correct.
On the other hand, asked to comment on the situation that would arise if a patient who had been taking Vioxx for a time discontinued doing so, Prof Celermajer said that there would be –
… almost immediate reversion of risk to normal because prostacyclin is regenerated, wherever it’s coming from, within 24 hours of stopping the drug. So the FitzGerald imbalance comes on in 24 hours and the FitzGerald imbalance goes within 24 hours.
I have set out Prof Harper’s view on this subject at para 542 above.
550 As Prof Zipes pointed out, the debate here involves the question whether Vioxx had not merely an acute effect in facilitating thrombosis but also a longer-term effect of promoting the development of atherosclerosis. For reasons explained earlier, I am not satisfied that Vioxx did have the latter effect. I note also that the follow-up data from the APPROVe trial, to the extent that it related only to the follow-up period as such, showed no statistically significant relative risk of taking Vioxx rather than placebo: see the third table in para 196 above. The conclusions which I reach as to the existence of a plausible biological mechanism for the risk disclosed by the main body of data in that trial, therefore, must be confined to the circumstances of a patient who is taking Vioxx more or less continuously. I deliberately express this conclusion in general terms because I wish neither to anticipate the circumstances of any particular group member in the present case, nor to prejudge the question whether his or her actual pattern of consumption of Vioxx was such as would provide a proper basis to infer the existence of the association for which the applicant contends.
Susceptibility of the atherosclerotic vasculature
551 An important aspect of the applicant’s case was that the risk of taking Vioxx was especially elevated in patients with existing atherosclerosis. At the mechanistic level, that conclusion was said to flow from an understanding of the consequences of the inhibition of COX-2 in the vasculature. Even allowing for the rejection of Prof Zipes’ view that Vioxx accelerated atherosclerosis and destabilised plaque, the post‑rupture pro‑thrombotic impact of the inhibition of COX-2 necessarily meant that it was patients who were susceptible to plaque rupture (ie those at a relatively advanced stage of atherosclerosis) who stood the greatest risk of the development of thrombosis. This was the position for which Profs Zipes and Harper contended in the joint cardiology report:
In our opinion, it is less likely that Vioxx would have an adverse effect on a normal artery. An atherosclerotic substrate is likely to be a necessary prerequisite for the adverse effects of Vioxx to occur. Therefore patients with known atherosclerosis (such as patients who have had previous heart attacks) or patients with multiple risk factors and therefore a high likelihood of clinically silent atherosclerosis are more likely to develop adverse effects from Vioxx therapy than a normal healthy population. To support this contention in both the VIGOR and the follow up of the APPROVe study, patients with known atherosclerosis or patients at high risk of atherosclerosis had a higher risk of adverse cardiovascular events than the remainder of the patient population.
552 This general topic was the subject of considerable controversy as between the cardiologists in their concurrent session. Speaking of himself and Prof Vaughan, Prof Celermajer said:
[W]e are of the strong belief that there is so much happening in the vicinity of a plaque rupture with other things that predispose to clotting such as tissue factor, plasminogen activator inhibitor 1, thromboxane particularly … [t]here is an enormous amount of that and that Vioxx compared to what’s going on at that local site we biologically don’t find it plausible that … COX-2 inhibition would influence that process.
Likewise, Prof Vaughan said:
Once a plaque ruptures, collagen is exposed in the blood vessel wall that initiates thrombosis. That collagen is the powerful driver of clot formation, that collagen promotes the activation of thrombin and that collagen also promotes the activation of platelets. Vioxx has nothing to do with the exposure of that collagen, the activation of tissue factor and the local activation of thrombin and those are the drivers of thrombosis in an acute coronary syndrome.
Prof Zipes’ response on this aspect was:
Plaque rupture is a major mechanistic foundation for heart attacks and very likely there are silent plaque ruptures. A very good pathologist in the States has shown the arteries of people who have died with heart attacks, that there is the evidence of plaque rupture healing over, plaque rupture healing over, three and four layers of this before finally the material within the plaque is extruded into the blood vessel and causes the major clotting that Professor Vaughan alluded to. I certainly agree with that. What we don’t know is with the rupture, heal, rupture, heal, how do we know that that healing process was not mitigated by prostacyclin? It’s certainly possible that it’s playing a reparative role until at one point it becomes overwhelmed, a big thrombus develops and plugs up the artery. That’s as reasonable an hypothesis as any other.
Prof Harper noted that most plaque ruptures were clinically silent, and “heal up”. But he added that, if a plaque rupture occurs, in the presence of Vioxx it was more likely that the plaque rupture would lead to a clinical event than if Vioxx were not present. The amount of platelet aggregation and clot formation would be greater in the presence of Vioxx than it otherwise would have been. Prof Harper went on to discuss the circumstances of the applicant, and I shall return to that in a later section of these reasons.
553 Prof Celermajer interpreted the evidence of Prof Zipes and Harper to which I have just referred as involving “two very interesting conjectures”, the second of which was Prof Harper’s “hypothesis” that (as articulated by Prof Celermajer) “prostacyclin in the local area of a plaque, if not inhibited, is sufficient to overcome the thrombotic effects of plaque rupture”. Prof Celermajer regarded that as “an extraordinary assertion that absent prostacyclin someone is much more likely to thrombose on the top of a large ruptured plaque”. He described it as a “mechanistic conjecture without any supporting data”. He referred to a published paper which reported on a study in which a high dose of prostacyclin – considerably in excess of what exists physiologically – was infused into occluded thrombosed coronary arteries of humans. Even that was insufficient to break the thrombi that had formed in the occluded arteries. Prof Zipes responded that prostacyclin was not a thrombolytic agent, it was a platelet inhibitor which would work by inhibiting the platelet from developing in the first place.
554 In the course of this debate, Prof Zipes gave the following evidence:
[T]here are numerous data indicating that the individual at risk, because of abnormal coronary arteries, is at greater risk from COX-2 inhibition, regardless of the mechanism. … The diabetics in APPROVe have a higher relative risk. The hypertensives in APPROVe had a higher relative risk. That increase in risk holds true in virtually all – certainly the controlled trials and in many of the epidemiologic or meta‑analyses. As an example, the most recent publication by Baron on the long term evaluation of the APPROVe patients makes the statement very clearly, that those at higher cardiovascular risk had a greater effect from the COX-2 inhibition.
This evidence related to what Profs Zipes and Harper had said in the joint report, referred to at para 551 above. Giving detail to that opinion, the applicant pointed out that, in the VIGOR trial, the relative risk (of a CVT event) from taking naproxen (rather than Vioxx) was, for aspirin‑indicated patients, 0.20 (0.06, 0.71) and, for other patients, 0.42 (0.25, 0.72). As noted elsewhere in these reasons, 38% of the myocardial infarctions occurred in the 4% of the patients who were aspirin‑indicated. All of those (eight in number) occurred in the Vioxx arm. The applicant relied also on the APPROVe trial, in which the relative risk of encountering a CVT event for patients taking Vioxx rather than placebo was, amongst patients who had a history of symptomatic atherosclerotic cardiovascular disease, 9.59 (1.36, 416.36), and amongst other patients, 1.58 (0.95, 2.64). The APPROVe figures are, of course, of more utility for present purposes than those from VIGOR, because the latter leave open the interpretation that it was naproxen that was especially cardioprotective for aspirin‑indicated patients, rather than that Vioxx was especially harmful to them.
555 The respondents might well have embraced the APPROVe data as confounding any suggestion that, for patients without a history of symptomatic atherosclerotic cardiovascular disease, Vioxx was associated with an elevated risk of CVT events. However, they did not take that approach. Instead, they resisted any suggestion that it was especially amongst patients with existing atherosclerosis that the consumption of Vioxx led to an increased risk of CVT events. The respondents submitted that the applicant’s use of data from VIGOR and APPROVe with respect to patients with existing atherosclerosis was a “post‑hoc subgroup analysis” which was neither meaningful nor reliable. They relied on the evidence of Prof Celermajer, who said, in his report, that it was important to avoid “post‑hoc analysis of non‑prespecified subgroups (meaning one should not look back at the data and invent new questions after having knowledge of the results)”. He gave as an example what he described as “the most infamous illustration” of such a mistake, namely, that of a study in which it was found (post‑hoc) that the administration of aspirin was beneficial for survivors of acute myocardial infarction, except in the case of patients born under the signs of Libra and Gemini. He said that post‑hoc subgroup analyses were “an unwise thing to do in any clinical trial other than to generate hypotheses which later need to be tested”. Better quality data, in Prof Celermajer’s view, were provided in the pooled analysis provided by Merck to the FDA on 22 March 2004, in which, amongst “high risk” patients, the relative risk of encountering an APTC endpoint from taking Vioxx rather than placebo was 1.36 (0.78, 2.37). This reflected what he and Prof Vaughan said in the joint cardiology report:
Regarding “high risk groups”, we note the pooled analysis (FDA 2004, Table 25[)], of APTC endpoints; RR 1.36, 95% CI 0.78 – 2.37 and we believe these data are more reliable than post hoc subgroup analyses.
556 In the 2004 pooled analysis to which Prof Celermajer referred, the relative risk (of encountering an APTC endpoint), for patients not in the high‑risk group, was 0.94 (0.54, 1.61). The subgroup‑by‑treatment interaction test revealed a significant difference only where p < 0.3187. This was well outside even the most liberal cut point mentioned by Dr Marks for tests of interaction (see para 335 above). The analysis did not set out corresponding results in the case of CVT events. By contrast, the Clinical Study Report for the APPROVe trial did set out the tests for interaction as between subgroups in relation to CVT events. Here the corresponding figure was 0.096. Although higher than the conventional figure for statistical significance (p < 0.05), this figure is comfortably within Dr Marks’ more liberal cut point for tests of interaction. Also, for those taking Vioxx with such a history, the risk of a CVT event was 3.82 (1.83, 7.02), while for those taking Vioxx without such a history, the corresponding figure was 1.29 (0.90, 1.78). These are 95% confidence intervals, and there is no overlap.
557 I am of the view, with respect, that Prof Celermajer’s rejection of the utility of the data from clinical trials that tend to show a stronger association between the inhibition of COX-2 and CVT events in patients with existing atherosclerosis was rather too categorical than the broad scope of the current inquiry justifies. It may be inappropriate to draw the kind of firm scientific conclusions as to non‑prespecified associations shown by the results of clinical trials that one would draw from the data which related to the prespecified endpoints of interest. But I consider that data of the former kind may be used – in a forensic if not a scientific setting – as a measure of the plausibility of the mechanism which, it is hypothesised, lies behind the excess of observed CVT events amongst patients in the Vioxx arms generally. If the FitzGerald hypothesis were valid, the existence of a relatively advanced atherosclerotic state would seem to be a necessary precondition to the occurrence of the events thereby hypothesised. For example, if the hypothesis were indeed the explanation for the relatively greater incidence of CVT events observed in APPROVe, one would expect to see that reflected particularly in the subgroup of patients who had a history of symptomatic atherosclerotic cardiovascular disease. That is exactly what one does see. So to observe is not to engage in a post‑hoc analysis of the kind rejected by the experts in this case. Rather, it is to observe that, if otherwise the evidence is such as to justify the conclusion, as urged by the applicant, that the FitzGerald hypothesis is a plausible explanation for the excess of CVT events seen in the Vioxx arm of APPROVe, the even more pronounced such excess observed in the subgroup presently of interest is consistent with that conclusion. It is not, of course, the only factor to be taken into account at this level, but it is a factor and it does add credibility to the mechanistic explanation for which the applicant contends.
558 In their written submissions in reply, the respondents advanced a number of other reasons why the applicant’s contention that Vioxx was particularly harmful in patients with existing atherosclerosis should not be accepted. The first related to the applicant’s own conformity with the so‑called high risk criteria in VIGOR and APPROVe. I shall return to that issue in a later section of these reasons. The second was that the identifier “patients with existing atherosclerosis” was “overly broad and vague”. I accept that, but it does not mean that some more specific linking of the circumstances of an individual with the extent of CVT risk posed by Vioxx may not be recognised, or is not implicit in the applicant’s submissions. The third was that the applicant was being selective in pointing to the sub‑groups of which he was a member by reference to an association with elevated risk. It was said that he ignored other subgroups of which he was a member (eg that of non‑diabetics) where there was no, or at least no elevated, risk. The respondents’ point here is, however, a distinction without a difference. In the passage set out at para 551 above, Profs Zipes and Harper said that patients with multiple risk factors had a high likelihood of having clinically silent atherosclerosis. If the existence of advanced atherosclerosis is established, it seems to me to be beside the point to say that a particular patient may not be a member of a different high risk group.
559 The respondents’ fourth reason involved the proposition that the evidence that Vioxx increased the risk in patients with risk factors was “weak and hypothesis generating at best”. It was said that the applicant had ignored “inherent imprecision and contradictory data”. As to the latter aspect, the respondents drew my attention to no “contradictory data”. As to the former, they pointed (without elaboration) to the following passage in Baron (2008):
Although our subgroup analyses suggest that individuals with risk factors for cardiovascular disease have higher rofecoxib relative risks than do healthy individuals, our data are not conclusive on that point. Tests for interaction were not significant, and the differences in rofecoxib risk between subgroups were compatible with chance.
This was, however, a general statement by the authors about the subgroup analyses, where the p‑values for interaction ranged from 0.013 to 0.081. In the case of the “high cardiovascular risk” subgroup, the relative risks of taking Vioxx (APTC endpoint) were 2.28 (1.25, 4.17) for those in that subgroup and 1.29 (0.70, 2.36) for those not in the subgroup. The p‑value for interaction was 0.19. Although this figure lies well outside the conventional limit for statistical significance, it is marginally within the most liberal of the cut points for the significance of interaction proposed by Dr Marks.
560 The risk figures taken from Baron may be compared with those relied on by the applicant, and referred to at para 554 above. The first difference is that the latter related to CVT events, while the Baron figures related to the APTC endpoint. Another, obvious, difference is that the figures on which the applicant relied were based on the APPROVe study period as such, whereas the Baron figures included data up to 31 October 2005. A third, and potentially important, difference is that, as stated above, the “high risk” group of patients in the figures on which the applicant relied were those with a history of symptomatic atherosclerotic cardiovascular disease, whereas the Baron “high risk” group included (in addition to patients with such a history) patients who had at least two of: history of hypertension, hypercholesterolemia or diabetes, or currently being a smoker. There were 380 and 342 such persons in the Vioxx and placebo arms of the study, respectively. In the group which yielded the figures on which the applicant relies, by contrast, there were only 116 and 102 patients with a history of symptomatic atherosclerotic cardiovascular disease in the Vioxx and placebo arms of the study, respectively. Most probably because of the greater numbers used in the Baron data, the confidence intervals are tighter. However, the impression given by both sets of figures is broadly the same: the relative risk of the endpoint of interest (from taking Vioxx) was unambiguously significant in the subgroup at high risk, and was greater than unity, but not significant at the 95% level, outside that subgroup.
561 With respect to the respondents, I think it would be naïve not to recognise that a history of symptomatic atherosclerotic cardiovascular disease is a circumstance of importance in the interpretation of the data yielded by the APPROVe study. Its importance lies not only in the association implied by the statistics, but in the support which it provides for the FitzGerald hypothesis, particularly as explained by Prof Harper. Returning to the question which I left unanswered in para 530 above, I am persuaded that that hypothesis does provide a plausible biological explanation for the excess of CVT events, and of myocardial infarctions particularly, observed in APPROVe.
562 I should make clear, however, that this conclusion does not involve a finding that the FitzGerald hypothesis is valid. Much less does it involve a finding that, in every person who took Vioxx, the mechanism proposed by the FitzGerald hypothesis was at work, day and night as it were, promoting thrombotic conditions in his or her cardiovascular system, and that any CVT event suffered by that person must necessarily have been contributed to by the drug. The conclusion I reach is no more than invited by the applicant, namely, that such a mechanism provides a plausible biological explanation for the results of APPROVe and the other relevant studies to which I have referred. I shall presently consider the omnibus question of whether Vioxx increased the risk of suffering the pleaded cardiovascular conditions, and if did so, that conclusion, and the cardiological evidence generally, including that which has caused me to come to a view about the FitzGerald hypothesis, will inform the next stage of the court’s task, namely, whether the consumption of Vioxx in fact contributed to the conditions suffered by the various group members, including the applicant.
Other Bradford Hill criteria
563 The second and fifth of the Bradford Hill criteria (temporality and reversibility respectively) played a minor part in the evidentiary controversy before the court. As to temporality, Prof Woodward noted that, in VIGOR, the subjects took Vioxx before their CVT events (and other outcomes) were observed, “thus showing that the postulated cause preceded the effect.” He added that the conclusion was strengthened by the exclusion of patients with a recent history of cardiovascular disease. Prof Celermajer’s comment was that temporality was “of little forensic assistance” since, in any clinical trial, any observed effect will follow the exposure, as observations only commence at the time of first exposure.
564 As to the fifth Bradford Hill criterion, Prof Woodward said that no research had looked at evidence of reversibility of effect, so that this criterion was neither indicated nor counter‑indicated. Prof Celermajer’s only comment on this was to point to the APPROVe extension data, to which I have referred in para 196 above. He said that, overall, the relative risk amongst the patients studied in the extension was “very similar” to that observed in the original study. The implication was, I take it, that, if Vioxx did have an effect on the incidence of myocardial infarctions (and CVT events generally), it was not reversible.
Other evidence of risk relied upon by the applicant
565 The applicant also relied upon indirect evidence of Vioxx being associated with CVT events. In the concurrent cardiology session, Prof Celermajer agreed that, in February 2005, the joint meeting of the Arthritis Advisory Committee and the Drug Safety and Risk Management Advisory Committee of the FDA unanimously expressed the view that the available data supported a conclusion that rofecoxib significantly increased the risk of cardiovascular events. Prof Celermajer pointed out that “cardiovascular events” in context included “heart failure” which, he noted, was not one of the pleaded cardiovascular conditions. Moreover, the issue upon which the joint meeting of the committees apparently expressed a view about rofecoxib was a central question in the present litigation. The court heard much direct, including expert, evidence on that question. An opinion expressed about it, outside court, by persons not called to give evidence should not be accorded much weight, even if Prof Celermajer’s evidence was not the subject of objection. Given the very detailed, and genereally sufficient, treatment of the subject by the witnesses actually called by the parties, I am not disposed to allow the view of these committees either to confirm or to qualify the conclusions which I would otherwise reach.
566 The applicant relied also upon Merck’s own actions in withdrawing Vioxx from the market on 30 September 2004. He submitted that this indicated Merck’s understanding that Vioxx caused an increased risk of CVT events. Although the forensic impact of this evidence cannot be doubted, ultimately I do not think it goes any of the distance towards establishing that Vioxx did lead to increase in such a risk. The results of the APPROVe trial speak for themselves. The way Merck reacted to them neither strengthens their scientific relevance nor excuses the court from considering the many issues that have been canvassed in this part of my reasons. Specifically, I do not consider that the respondents’ entitlement to put the applicant to his proof as to the relationship between the consumption of Vioxx and the occurrence of the pleaded cardiovascular conditions has been compromised by the commercial decision made by Merck on 30 September 2004.
Vioxx and the risk of myocardial infarction
567 To date, I have for the most part considered the association between the consumption of Vioxx and CVT events as a group. That was in part because the published and otherwise available research dealt with the problem along that axis and in part because such evidence of the association as it existed from time to time informed, or ought to have informed, the response of Merck as manufacturer of Vioxx. However, the question which the applicant’s allegation raises is whether the consumption of Vioxx increased the risk of each of the pleaded cardiovascular conditions considered as a separate entity. It is to this question that I now turn, commencing with myocardial infarction.
568 The general body of data to which I have referred above amply justifies the finding that, at the population level, the consumption of Vioxx increased the risk of myocardial infarction. Here I refer to my conclusion in para 476 and to the reasoning associated with it. Indeed, it was at times implicit in the respondents’ case that some of the more conspicuous CVT relative risk outcomes were driven by the myocardial infarction components of the general data. As to the mechanistic explanation for the excess of myocardial infarctions amongst patients taking Vioxx, I refer to, and adopt in the context of myocardial infarction specifically, my conclusion at para 561 above. I consider that the mechanism proposed by the FitzGerald hypothesis provides a plausible explanation for the myocardial infarction results of APPROVe. At the same time, it provides a rational justification for exercising caution in the application of population risk increase data to every individual. For reasons which I have attempted to explain, the hypothesis would provide a more obviously persuasive explanation for the increase in risk seen in APPROVe in the case of patients with symptomatic atherosclerotic cardiovascular disease than in the case of other patients.
569 As for the remaining Bradford Hill criteria, I would place the dose‑relationship at a somewhat lower level of importance, but, for reasons explained above, it does seem to be there, and is another factor in support of the applicant’s case on risk. The other criteria played very little part in the controversy between the experts in the case, but to the extent that they have any relevance, they appear to support the position for which the applicant contends. As obvious as it may sound, the fact is that exposure to Vioxx preceded the myocardial infarctions which were the subject of the trials and studies referred to in the evidence. Only in the setting of the VIGOR trial was there an alternative explanation with any claim to be convincing. Even then, as I have said, that explanation did not deal with the full extent of the relative risk revealed by the data. In other settings, the respondents did proffer some alternative explanations for consideration, but it was not their case that any of them should be regarded as convincing. I have dealt with them in the various contexts in which they were proffered in my reasons above. As to reversibility, I accept Prof Woodward’s evidence that there has been no useful research on that subject.
570 I find, therefore, that the consumption of Vioxx increased the risk of suffering myocardial infarction. It did so by a factor of about 2. In the present case, the applicant takes a further step and alleges that the consumption of Vioxx “materially” increased that risk. By that allegation the applicant meant that the increase was one to which a reasonable person would be likely to attach significance. The respondents resisted that proposition on the basis that the reaction of a reasonable person to a given increase in risk would depend entirely on what the level of risk was in the first place. If the original level of risk were miniscule, a doubling thereof would be of no significance to a reasonable person, according to the respondents.
571 In the legal (ie not the factual or analytical) submissions made on behalf of the applicant, counsel made clear from where the requirement of “materiality” derived. They relied upon the passage in Rogers v Whitaker (1992) 175 CLR 479, 490 to which I refer at para 784 below. I do not here propose to anticipate the issues to which I refer in that part of my reasons, but I note that their Honours in the High Court confined their connotation of the notion of materiality to “the circumstances of the particular case”, and to the thinking of “a reasonable person in the patient’s position”. In their legal submissions, counsel for the applicant seemed to embrace these limitations, but they paid no attention to the interface between their submissions as so made and the sweeping allegation in their client’s pleading that the consumption of Vioxx materially increased the risk of suffering the pleaded cardiovascular conditions. Counsel for the respondents paid considerable attention to that interface, and submitted that, because of the limitations expressed in Rogers v Whitaker, it was not possible to make good any broad proposition about the materiality of any increase in risk. All would depend upon the circumstances in which the individual patient was placed.
572 To the extent that the applicant’s allegation of materiality performs service in his negligence case specifically, I shall deal with it in Part VII of these reasons. However, its role is not so confined in his case generally. In the aspect with which I am presently dealing, the applicant makes a general allegation about risk, and asserts that the risk was such as a reasonable person would regard as significant. He asks the court so to find without consideration of the particular circumstances of any person. I should deal with the allegation in the context in which it is made. Here it is important to refer to another submission made by the applicant, namely:
In order to satisfy the “materiality” requirement it is not necessary to establish that the patient would have acted upon a warning if given – this is a question of causation. The test is “somewhat lower than that” and materiality will be established if the reasonable or particular person “would have been likely seriously to consider and weigh up the risk before reaching a decision”: Rosenberg v Percival (2001) 205 CLR 434, 459 [79], [80].
This is a fairly low threshold, and would readily be crossed where the event of which there was an increase in risk was a condition as potentially serious as myocardial infarction. In my view, whatever a particular patient’s base‑line of risk of myocardial infarction, a doubling thereof would be regarded as significant by the reasonable person and should, therefore, be regarded as material in the sense employed by the applicant.
Vioxx and the risk of thrombotic stroke
573 The next condition I propose to consider is thrombotic stroke, a subject which I briefly introduced in para 316 above. Although the applicant submitted that Vioxx increased the risk of CVT events, and thrombotic stroke was such an event, in his final written submissions no attempt was made to deal with the association between the consumption of Vioxx and the occurrence of thrombotic strokes as such. Those submissions noted the stroke data from some of the studies referred to in the evidence, but contained no reasoned argument about them. In their final written submissions, the respondents asserted that the applicant’s experts had not directly addressed the issue of risk related to the pleaded cardiovascular conditions other than myocardial infarction, and had not provided data to support the applicant’s contention of increased risk. In oral submissions, counsel for the respondents submitted that, as they read them, the applicant’s written submissions made no reference to the pleaded cardiovascular conditions other than myocardial infarction. They (counsel for the respondents) proceeded to deal with the evidence on thrombotic stroke. In their oral submissions (which followed), counsel for the applicant neither responded to these submissions made on behalf of the respondents, nor specifically argued the matter of the risk of thrombotic stroke. They did, however, file a memorandum in response to the respondents’ submissions, in which they said:
Merck argue that there [sic] no increased risk of other pleaded conditions. As noted previously, the CVT endpoint is the most useful endpoint in attempt to gain information about the pleaded conditions. The numbers are too small to provide useful information. Nevertheless, the APPROVe study showed a 3 fold increase in ischaemic strokes.
I take it that the “3 fold increase” mentioned here was a reference to the difference between 18 and 6 ischaemic cerebrovascular strokes reported in Baron (2008) in relation to the APPROVe data as extended to 31 October 2005. The same figure is given in the second table in para 196 above, where the relative risk of suffering such a stroke from taking Vioxx rather than placebo is stated as 3.08 (1.22, 7.76). In the study period as such, the relative risk was 1.99 (0.67, 6.55).
574 In the joint report of the cardiologists, Profs Celermajer and Vaughan said:
There are no data linking VIOXX consumption to thrombotic stroke. In fact, we believe there are compelling data to show that VIOXX does not predispose to thrombotic stroke. These data have been published by Kearney (2006; RR 1.02, 95% CI 0.71‑1.47) and Chen (2007; RR 1.06, 95% CI 0.66‑1.72). Further, we note the data tabulated in Marks’ Appendix B of the Biostatistician’s joint report (2009, RR for thrombotic stroke 0.67, 95%CI 0.42‑1.09).
What Profs Celermajer and Vaughan there said about Kearney (2006) was correct, although it may be added that that study was a meta‑analysis (relevantly to strokes) of 37 trials, and it does not appear that the strokes covered by the studies analysed were limited to thrombotic ones. The specific figure for Vioxx was not given, but, from a graph in the article, it is apparent that the figure was very close to unity. There were 34 strokes amongst 6638 patients taking Vioxx, and 34 strokes amongst 6415 patients taking placebo.
575 Although they were not cross‑examined on the subject, I think that Profs Celermajer and Vaughan were not correct in what they said about Chen (2007). Chen (2007) was a meta‑analysis which dealt only with myocardial infarction. It said nothing about strokes. It may be that the Professors intended a reference to an article published in 2008 by the same authors, and referred to in the list of sources appended to Prof Celermajer’s report, titled “Do selective COX-2 inhibitors increase the risk of cerebrovascular events? A meta‑analysis of randomised controlled trials”. However that may be, the fact is that Profs Celermajer and Vaughan gave evidence of a relative risk figure for strokes as found by Chen, and they were not challenged on that evidence.
576 Neither, I would add in passing, were Profs Celermajer and Vaughan challenged on their emphatic statement that there were no data linking the consumption of Vioxx to thrombotic stroke. They were not referred to the relative risk for stroke yielded by the extended APPROVe data, and reported in Baron (2008): 2.17 (0.98, 4.80); p = 0.05. These data included haemorrhagic strokes, of which there was one in the Vioxx arm and there were three in the placebo arm. I have referred to the numbers of ischaemic strokes above.
577 In an appendix to the biostatisticians’ joint report, Dr Marks set out the results of a number of studies and trials conducted by Merck to the extent that they related to ischaemic cerebrovascular strokes. He calculated an overall relative risk figure for taking Vioxx rather than the comparator (which was not always placebo): 0.67 (0.42, 1.09). The figure included from the APPROVe data was 2.00 (0.74, 5.40), which Dr Marks said was sourced in Merck’s background information for the FDA Advisory Committee dated 21 January 2005. That document is in evidence, and I must say that it does not appear to contain any such figure. It does show that, in the APPROVe data as originally prepared (ie counting events up to the 14th day after discontinuation of treatment), there were 11 ischaemic cerebrovascular strokes in 3041 patient‑years in the Vioxx arm, and 6 such events in 3315 patient‑years in the placebo arm. The document also made the following statement:
Treatment with rofecoxib was associated with an overall relative risk of 1.96 (95% CI: 1.20, 3.19) compared to placebo for the development of confirmed thrombotic cardiovascular serious adverse events, due primarily to a higher incidence of acute myocardial infarction and ischemic cardiovascular stroke.
As I have indicated above, the relative risk figure disclosed in the Clinical Study Report of 6 June 2005 was 1.99 (0.67, 6.55). Dr Marks did not refer to the figure given by Baron and colleagues. However these variations and differences may be, the applicant appeared to accept the accuracy of the overall risk figure given by Dr Marks in his appendix: he was not cross‑examined on it, and no mention was made of it in the submissions made on behalf of the applicant.
578 Expert evidence specifically related to strokes was given by Prof Graeme Hankey, called by the respondents. Prof Hankey is a consultant neurologist and head of the stroke unit at the Department of Neurology at Royal Perth Hospital, and a Clinical Professor at the School of Medicine and Pharmacology at the University of Western Australia. He considered that the best available evidence for any association between exposure to Vioxx (or other coxibs or NSAIDs) and ischaemic stroke (or transient ischaemic attack) was Kearney (2006). That study revealed that, by comparison with taking placebo, the relative risk of stroke from taking a coxib was 1.02 (0.71, 1.47). Prof Hankey concluded from this that there was no evidence that exposure to a selective COX-2 inhibitor caused stroke. He also noted the conclusion of the authors of the Kearney article that there was “complete consistency” (ie no evidence of any heterogeneity) as between the different coxibs studied. Under cross‑examination, it was put to Prof Hankey that the Kearney results did not make a distinction between ischaemic strokes and haemorrhagic strokes. His response was that that “generally doesn’t matter because 85% of strokes are ischaemic”. Notwithstanding that, he accepted that, if one’s object were to determine the actual effect of a trial drug, one would want to know where in the study the haemorrhagic strokes occurred and where the ischaemic strokes occurred.
579 Prof Hankey was taken to the stroke results yielded by the extended APPROVe data, as reported in Baron (2008). He pointed out that the overall stroke risk figure of 2.17 was not statistically significant at the 95% confidence level. He agreed that the confidence interval represented a bell‑shaped curve, the lower tail of which just crossed unity. He accepted that the position would be different if, say, a 90% confidence interval were employed. Prof Hankey’s other concern with the Baron article was that, after discontinuing study treatment, 311 patients in the Vioxx arm and 324 patients in the placebo arm had been unblinded at their own request. By then, there had been considerable publicity given to the international withdrawal of Vioxx, and to the reasons for it. Prof Hankey considered that there was a risk that some investigators (the treating practitioners) might, as a result, be more ready to recognise particular symptoms as a stroke in patients known to have taken Vioxx than in patients known to have taken placebo. Notwithstanding those reservations, in his report Prof Hankey said that he suspected that the results reported in Baron (2008) were nevertheless valid.
580 In his cross‑examination of Prof Hankey, senior counsel for the applicant pointed out that, in the Vioxx arm of APPROVe (extended data), there had been 18 ischaemic strokes and only one haemorrhagic stroke. This pattern of distribution was said to be rather different from the distribution found in the population generally which, as Prof Hankey had said, showed that about 85% of strokes were ischaemic. By contrast, in the placebo arm, one half of the strokes (three compared with six) were haemorrhagic. It was when these and similar figures from Baron (2008) were being canvassed in the course of his oral evidence that Prof Hankey expressed substantial doubts as to the validity of the Baron results, on account of the potential for bias resulting from unblinding in the way I have described. He was disposed to think that the original results (ie counting only events which occurred up to the fourteenth day after discontinuation) were the more reliable. Those results showed that, in the Vioxx arm, there were 11 ischaemic strokes and one haemorrhagic stroke and, in the placebo arm, there were six ischaemic strokes and two haemorrhagic strokes. However, whichever data were relied on, Prof Hankey thought that the individual relative risk figures for ischaemic and haemorrhagic stroke (which had not been calculated by Baron and colleagues) would lie within very wide confidence intervals and, inferentially at least, would be of little utility. He was not referred to, and I do not know what he would have made of, the data in Merck’s extended analysis to which I have referred in para 195 above.
581 Under re‑examination, Prof Hankey was taken to Merck’s background information for the FDA Advisory Committee dated 21 January 2005. His attention was drawn to a table of confirmed thrombotic cardiovascular serious adverse experiences in VIGOR. For ischaemic cerebrovascular stroke, the table showed that there had been nine events in 2697 patient years in the Vioxx arm, and eight events in 2698 patient years in the naproxen arm. He said that these data were “absolutely in line with the meta‑analysis of Kearney”. His attention was drawn to a table in the same report which summarised the thrombotic cardiovascular serious adverse experiences in Protocols 078 and 091 (the Alzheimer’s disease data). For ischaemic cerebrovascular stroke, there were six events in 1661 patient-years in the Vioxx arm and 17 events in 1917 patient-years in the placebo arm. Prof Hankey said that those figures did not cause him to change the opinions which he had expressed. He noted that here there was a trend in the other direction, suggesting that Vioxx prevented ischaemic stroke, but the data involved “very unstable numbers” for which there would be wide confidence intervals, and a relative risk figure (had it been calculated) that probably would not be statistically significant.
582 A striking characteristic of the table extracted from Kearney (2006) set out at para 427 above is the different relative risk patterns disclosed in relation to myocardial infarction and stroke. Much of the cross‑examination of Prof Hankey was concerned to demonstrate that, if the consumption of Vioxx was shown to be associated with an increased risk of myocardial infarction, one ought to be able to conclude that there was likewise an increased risk of thrombotic stroke. Prof Hankey seemed to accept the logic of that proposition, but only up to a point, since, as he said, the relationship between thrombotic occurrences in the vasculature and stroke was somewhat more complex than the relationship between such occurrences and myocardial infarctions. However, I did not understand the applicant ultimately to submit that I should find, contrary to the published research, and without any unequivocal affirmation by any of the expert witnesses, that the consumption of Vioxx was most probably associated with an increased risk of stroke for no reason other than that it was shown to have been associated with an increased risk of myocardial infarction and, as a matter of logic, a like conclusion ought to be drawn in the case of strokes.
583 On the evidence, I am not satisfied that the consumption of Vioxx gave rise to an increased risk of thrombotic stroke.
Vioxx and the risk of the other pleaded cardiovascular conditions
584 As to the other pleaded cardiovascular conditions (unstable angina, transient ischaemic attack and peripheral vascular disease) there was no evidence from which I could find that an increased risk of any of them arose from the consumption of Vioxx. I have referred to the opinion of the cardiologists at para 317 above. The only direct submission made by the applicant with respect to these conditions was that which I have set out at para 573 above. I could not accept the applicant’s invitation to transfer the established risk of CVT events generally to each of the pleaded cardiovascular conditions, including this final group of three. As I have said elsewhere, the conclusion that the consumption of Vioxx led to an increased risk of suffering CVT events generally does not mean that there was an increased risk of suffering each of the pleaded conditions, considered as a separate entity. Only in relation to myocardial infarction have I found that such an increased risk probably existed. In relation to the other conditions, I am unable to make any such finding.
part v − what the respondents knew or ought to have known
585 In his Further Amended Statement of Claim, the applicant alleged that, at all material times after 30 June 1999, the respondents knew or ought to have known that the consumption of rofecoxib materially increased the risk of suffering the pleaded cardiovascular conditions. I have held that the only condition in relation to which there was in fact a material increase in risk is myocardial infarction, but the present question is not confined to whether the respondents knew or ought to have known that the consumption of Vioxx materially increased the risk of suffering that condition. If there were information actually or constructively known to the respondents from which it ought reasonably have been concluded by them that the consumption of rofecoxib materially increased the risk of suffering cardiovascular conditions generally, that circumstance would also be relevant to the question whether the respondents discharged their duty of care to avoid exposing users of Vioxx to the risk of myocardial infarction. As has already been shown, the data which were available to the respondents down to September 2004 generally spoke in terms of a basket of conditions – be they CVT events generally or the conditions encompassed by the APTC endpoint – and they sometimes, but not invariably, provided information as to particular events, such as myocardial infarction, as well. It was implicit in the cases of both parties that it was appropriate for the respondents to have had regard to all the available data.
586 It is important, in an understanding of the applicant’s case in this regard, to note that the primary evidence upon which he relies to contend for a finding that the consumption of Vioxx in fact increased the risk of CVT events including myocardial infarction is the results of the APPROVe trial. Those results were not available, even in summary form, until about the end of September 2004. This raises the question whether the applicant is in any position to assert that Merck should, at some previous time, have reached the conclusion which he now invites the court to reach. Should Merck, operating within the milieu of the emerging facts and the developing science, have been more prescient than the applicant himself is now, with the added benefit of all the subsequent research and analysis (including the re‑analyses of Profs Madigan and Woodward) upon which he has relied in this case? On the final day of the hearing, I raised this problem with senior counsel for the applicant, during his final address. He said:
There’s two ways of answering that. The first is that the “ought to have known” is really a rolled up way of saying that [Merck] had warning signs which they should have investigated and if they had investigated they would have found the fact and so we attribute to them knowledge of the fact that they closed their eyes to. That analysis is directly applicable here, in our submission. Once they saw the warning signs we would say from the [osteoarthritis] and [rheumatoid arthritis] studies which were available to them by 2000 but certainly when they got the VIGOR results there was a clear warning sign which should have led them to investigate the fact.
It was submitted that the investigations which Merck should have undertaken were either or both of a cardiovascular outcomes study as to the effect of Vioxx and a study of the cardioprotective benefits of naproxen. Recognising that either of those experiments would take time, counsel submitted that the respondents should not have been marketing Vioxx in the meantime, at least without more conspicuous and categorical warnings than in fact were given.
587 The applicant’s case did, therefore, involve (in the alternative, it seems) the proposition that, even if the respondents were not fixed with actual or constructive knowledge that Vioxx increased the risk of suffering the pleaded cardiovascular conditions, at least they ought, acting reasonably, to have recognised the “warning signs” which were actually or constructively apparent to them. They ought to have concluded that, in a situation in which the science was evolving, there was a “risk” that it would ultimately be demonstrated that Vioxx did in fact increase the risk of suffering the pleaded cardiovascular conditions: “a risk that it increased the risk” as counsel for the applicant put it. This submission essentially involves a consideration of the credibility and strength of the “warning signs” actually or constructively apparent to the respondents over the course of the period between the release of the VIGOR results and the withdrawal of Vioxx from the market, and of the nature of the obligation of reasonable action imposed upon the respondents by the law of negligence in all of the circumstances then confronting them.
Before the results of VIGOR were available
588 It was submitted on behalf of the applicant that, even before the VIGOR trial, Merck was well aware of the FitzGerald hypothesis and was well aware, therefore, of the possibility that selective COX-2 inhibition might upset the balance between prostacyclin and thromboxane in the vasculature and lead to thrombotic outcomes. I accept that submission, but I also hold that it was not until Merck received the results of Protocol 023 some time after 29 August 1997 that the prospect of such outcomes was known, or could have been known, to Merck. Until then, it does not appear to have been suggested, either in the studies conducted by Merck or in the literature generally, that endothelial prostacyclin was synthesised by COX-2 rather than by COX-1. Indeed, it was Protocol 023 itself, one of Merck’s own studies, that first provided the pharmacological data by reference to which Dr FitzGerald articulated his hypothesis. Merck’s response to the results of Protocol 023 was to examine, so far as the data permitted, the occurrence of cardiovascular serious adverse events in its Phase IIb/III osteoarthritis trials. Those data, subject to limitations though they were, provided no signal for concern. Merck also referred the results to its Board of Scientific Advisers who, in May 1998, gave earnest consideration to the possibilities presented by the FitzGerald hypothesis in the context of the imminent commercialisation of Vioxx. They recommended the analysis of coronary events in existing and forthcoming trials of Vioxx. They considered that the subject of their deliberations (which included the possible cardiovascular benefits of Vioxx) were “part of the scientific intelligence gathering appropriate to the development of any truly novel pharmacological entity”. They took the view that there was “a strong mandate for introduction of Vioxx into medical practice as soon as is feasible”. Consistently with the recommendations of the board, Merck set about to develop the CV SOP.
589 Tendered without objection by the applicant was an executive summary of the deliberations of the “International Consensus Meeting on the Mode of Action of COX-2 Inhibition” held on 5‑6 December 1997. The only witness who referred to this summary was Prof Madigan, a biostatistician called by the applicant. In his report, he said:
The possibility of cardiovascular side effects of Vioxx was well established early on. In December 1997, the International Consensus Meeting on the Mode of Action of COX‑2 Inhibition considered adverse events. The executive summary stated, “although gastric intolerance was the most obvious [adverse event], renal and cardiac toxicity were important and a probable link between these two phenomena had been established. It is therefore important to monitor cardiac side effects with selective COX‑2 inhibitors.”
In his final submissions, the applicant relied upon this evidence. However, the passage in the executive summary to which Prof Madigan referred appeared under the heading “Traditional NSAIDs: defining the problem”. The full text of the paragraph relied on by Prof Madigan was as follows:
It was emphasized that the major reason for both the development of COX-2 inhibitors, and this meeting, was adverse events associated with NSAlDs. Although gastric intolerance was the most obvious, renal and cardiac toxicity were important and a probable link between these phenomena had been established. It is therefore important to monitor cardiac side effects with selective COX-2 inhibitors.
It is quite clear that, when the meeting noted that a probable link between gastric intolerance and renal and cardiac toxicity had been established, it was referring to the effect of traditional NSAIDs. There was no suggestion of any “cardiovascular side effects of Vioxx”, or of coxibs generally, as such. Indeed, the striking thing about the executive summary when read as a whole is that it makes no reference to the possibility of COX-2 inhibition having thrombotic consequences. Neither was there any reference to the results of Protocol 023. (I do not imply that there need have been, since they had not by then been published.)
590 The cardiologists called by both sides gave evidence which is important for this aspect of the applicant’s submissions. Having first been asked whether the FitzGerald hypothesis, accelerating atherosclerosis or destabilising atherosclerotic plaque was a physiological mechanism by which Vioxx increased the risk of suffering one of the pleaded cardiovascular conditions, they were then asked to state, with respect to any such mechanism as they affirmatively identified, what was the earliest date at which scientific knowledge enabled that conclusion to be reached. They said:
The Fitzgerald hypothesis was known to Merck in 1998. At this point in time, however, the risk (regarding the Fitzgerald hypothesis) was theoretical and there were other plausible mechanisms whereby Vioxx could potentially have a cardioprotective effect. For example, in 1998 there was a hope that anti‑inflammatory drugs such as Vioxx might actually protect against adverse cardiovascular events.
The cardiologists were then asked what was the earliest date at which scientific knowledge suggested that there was “a real (not fanciful) possibility” of such a conclusion being reached. Here they disagreed in respects to which I shall turn, but none suggested that there was such a possibility at any time before the results of the VIGOR trial were known.
591 There was one other event (or, more accurately, pair of events) to which the applicant pointed as demonstrating Merck’s awareness of the mechanism proposed by the FitzGerald hypothesis in 1999 or thereabouts. In written submissions filed on behalf of the applicant, it was said that, on 9 March 1999, one Dr Steven Nichtberger on behalf of Merck had filed an application for an international patent, under the Patent Co‑Operation Treaty, for an invention called “Combination Therapy and Composition for Acute Coronary Ischemic Syndrome and Related Conditions”. The abstract in the application read as follows:
The invention provides a method for treating, preventing, or reducing the risk of developing a condition selected from the group consisting of acute coronary ischemic syndrome, thrombosis, thromboembolism, thrombotic occlusion and reocclusion, restenosis, transient ischemic attack, and first or subsequent thrombotic stroke, in a patient, comprising administering to the patient a therapeutically effective amount of an antiplatelet agent in combination with a therapeutically effective amount of a COX-2 inhibitor. The invention also provides a pharmaceutical composition comprising a therapeutically effective amount of a COX-2 inhibitor, or a pharmaceutically acceptable salt thereof, and an antiplatelet agent, or a pharmaceutically acceptable salt thereof.
The applicant noted that, three days later, Dr Nichtberger filed an application for a US patent with an identical abstract. That was, of course, the same invention. These documents were produced to Dr Reicin – apparently without notice – during cross‑examination. She knew nothing of the applications. In re‑examination, having been given the opportunity to read a paragraph from the complete specification of the US patent, Dr Reicin said that the invention was for a product to treat patients at risk of recurrent thrombosis, with the hope that a combination of an antiplatelet inhibitor and a COX-2 inhibitor might be better in reducing the risk of thrombosis than either of them alone. She said that the invention was for a cardioprotective combination product. It was not for patients with arthritis. As though this evidence had never been given, it was submitted on behalf of the applicant that these patent applications suggested that Merck was concerned about the development of thrombotic events in at risk patients who were being administered COX-2 inhibitors. Whatever concerns Merck may have had, however, they were clearly unrelated to these applications. I need say nothing more about them.
592 For the above reasons, I reject the allegation that, prior to the release of the VIGOR results, the respondents knew or ought to have known that the consumption of Vioxx increased the risk of suffering the pleaded cardiovascular conditions.
Merck’s interpretation of the VIGOR results
593 The CV SOP was applied to the VIGOR trial. The results of that trial itself were relied on by the applicant to justify the conclusion that Merck knew or ought to have known that Vioxx increased the risk of myocardial infarction and of CVT events generally. Here I return to the cardiologists’ opinions as to the earliest date at which scientific knowledge was such as to justify the conclusion that there was a real (ie not fanciful) possibility that Vioxx increased the risk of the pleaded cardiovascular conditions. In this respect they all joined in the following opinion as to the position that was brought about by the availability of the VIGOR results:
We all recognise the potentially worrisome and potentially important signal of cardiovascular risk associated with Vioxx from VIGOR and believe that it mandated comprehensive analysis and investigation.
The sense of what the cardiologists meant would, I consider, be better conveyed by saying that the VIGOR results represented a worrisome and important signal of potential cardiovascular risk associated with Vioxx. It was a signal which mandated comprehensive analysis and investigation. It is in this sense that I shall read this passage in the cardiologists’ joint report.
594 Of the VIGOR trial, the cardiologists said that “cardiovascular events were monitored but were not pre‑specified as primary or secondary end points.” Thus, according to Profs Harper, Celermajer and Vaughan, “when the VIGOR data on cardiovascular events became available it was reasonable to regard these data as hypothesis generating.” Prof Zipes disagreed with the term “hypothesis generating”, because he believed that there were significant other data that validated the adverse cardiovascular signal. In his view, by at least the end of 2000 Merck had sufficient data “to realise that Vioxx had significant cardiovascular toxicity”. However, when, in the cardiology concurrent session, Prof Zipes was asked to identify those data, he referred to Prof Madigan’s re‑analysis of the Merck arthritis data (with which I have dealt earlier in these reasons), to an internal analysis by Dr Chen which was in fact undertaken in 2001, to Juni (2004) – mistakenly supposing that to have been published in 2000 – and to another internal analysis undertaken by Dr Shapiro in October 2000 which, to my reading of it, was consistent with the pooled analysis later to be forwarded by Merck to the FDA in January 2001. I consider that, in these circumstances, I am justified in finding that the better view, from a cardiological perspective, was that the VIGOR results presented a worrisome and important signal of potential cardiovascular risk associated with Vioxx, but did not justify the conclusion that the risk existed as a matter of reality rather than of hypothesis.
595 The applicant’s case against Merck, apropos the position in which it found itself in March 2000 and thereafter, was not confined to the allegation that it ought then to have been realised that a cardiovascular risk existed. It was submitted on his behalf that VIGOR delivered a signal to Merck that was both strong and, in the light of the then topical FitzGerald hypothesis, unsurprising, yet “Merck not only disregarded that signal, it acted as though no such signal existed”. In my view, these criticisms of Merck are unduly harsh, and not justified by the evidence. In the period immediately after the release of the VIGOR results, Dr Reicin and the other scientists at MRL worked very hard to understand those results. Indeed, Dr Scolnick, the head of MRL, immediately made the connection for which the applicant would contend – that the explanation may well lie in the results of Protocol 023 and in the cases referred to by Dr Oates in his letter of 13 August 1999. Because VIGOR was not a placebo‑controlled trial, immediately to have said that Vioxx was pro‑thrombotic would, in my view, have been a simplistic and inappropriate conclusion. As a matter of logic, the results might have reflected the pro‑thrombotic tendency of Vioxx, the cardioprotective work of naproxen, a combination of the two, or the play of chance. Aside from the play of chance (which Dr Reicin soon discarded as an explanation) there were three possibilities. The applicant criticised Merck for coming down decisively, and at an early stage, in favour of the cardioprotective work of naproxen as an explanation for the results. A more measured and responsible approach would have been to recognise the other two possibilities. There is force in that criticism, but by no means could it be said that Merck disregarded the signal given by VIGOR, or acted as though it did not exist.
596 It was submitted on behalf of the applicant that, at the time of the release of the VIGOR results, “no studies showed a cardioprotective effect of naproxen”. In their joint report, the biostatisticians agreed that “at the time of the VIGOR study, there was no extant evidence that naproxen was cardioprotective”. In their oral evidence, they made clear that the “time” to which they were referring was March 2000. They also made clear that, by “extant evidence” they intended a reference to evidence which appeared in the published literature.
597 Protocol 061 had not been published as at March 2000 (it was soon to be published as Van Hecken (2000) – Dr Gertz was one of the authors). As indicated above, that study was critical in Dr Reicin’s conclusion that naproxen was cardioprotective. The applicant submitted that Protocol 061 showed no such thing. He based that submission on certain propositions. First, Protocol 061 was a pharmacological study, not an RCT. Secondly, Protocol 061 looked at bleeding times in only eight young, healthy, women and, according to the applicant, “bleeding time studies are unreliable”. Further, there were important differences between the pharmacological effects of a drug on healthy arteries and the like effects on atherosclerotic arteries. Thirdly, Protocol 061, according to the applicant, demonstrated only “what Merck already knew”, namely, that some traditional NSAIDs had antiplatelet effects. Such effects “do not necessarily equate to cardioprotective effects”. Fourthly, aspirin was not a comparator in Protocol 061 and it was not, therefore, open to Dr Reicin to conclude that naproxen’s antiplatelet effects were similar to those of aspirin.
598 It is true that Protocol 061 was a pharmacological study rather than an RCT. The passage in the transcript of the cross‑examination of Dr Reicin to which the applicant referred in his submissions in this respect is unrevealing. I suppose that an RCT would have enabled scientists and researchers to draw a conclusion as to the association between the consumption of naproxen and particular clinical outcomes. However, I do not accept that the relevance of Protocol 061 to the exercise in which Dr Reicin and her colleagues were involved in March 2000 is to be depreciated by reason only that it was not a study of that kind. Those scientists were concerned to understand the effect of naproxen in the vasculature. Necessarily, they could work only with the material that was available to them. Perhaps fortuitously (because Protocol 061 was a Merck study), Dr Reicin had available to her unpublished material which threw important light upon the problem with which she was grappling. She is not to be criticised for having availed herself of that resource.
599 The only witness who gave evidence that bleeding time studies were “unreliable” was Prof Cleland, and that evidence was not given in the context of Protocol 061. In his report, Prof Zipes referred to the protocol, but did not suggest that the results were of little utility by reason of the circumstance that it was a bleeding time study. Neither was that proposition put to Dr Reicin or, for that matter, to any of the cardiologists. I could not hold, therefore, that, in March 2000, Dr Reicin, acting reasonably, ought to have discounted or depreciated the utility of the results of Protocol 061 by reason of the fact that it was a bleeding time study.
600 Neither was it put to any of the cardiologists that the results of Protocol 061 should have been discounted by Dr Reicin because the study involved healthy, rather than atherosclerotic, arteries. The evidence upon which the applicant relied in support of its submission that there were “important differences” between arteries in these states related to a different point altogether, namely, whether an atherosclerotic substrate was required for there to be increased cardiovascular risk from the consumption of Vioxx. There is no expert evidence in this case which would justify dismissing the significance of the results of Protocol 061 on the basis that the subjects had healthy, rather than atherosclerotic, arteries.
601 The difference between an antiplatelet effect and a cardioprotective effect of naproxen, in the context of Protocol 061, was referred to by Prof Zipes in his report:
Dr. Reicin fails to mention Protocol 061 also showed that all of the active agents, including naproxen, produced ≥65% inhibition of COX-2 activity, which reduced the excretion of PGI2 and PGE2 metabolites, the beneficial prostaglandins. This is what Vioxx does to increase the cardiovascular risk. Therefore, naproxen cannot be considered an aspirin substitute.
A similar distinction was made by Prof Harper in the passage which I have set out at para 369 above. As I have said in para 370, it seems that in due course it was demonstrated, at least in some studies, that the presumptive antiplatelet effect of naproxen likewise had cardioprotective consequences. However, this was not known in 2000. The applicant’s point was that, at that time, the research available to Dr Reicin did not justify her concluding that the antiplatelet effect of naproxen, such as it was, necessarily had those cardioprotective consequences.
602 While I consider that there is some substance in the point here being made by the applicant, his case is that Dr Reicin ought to have recognised the VIGOR results as a demonstration of what happens when the platelet‑aggregating functions of thromboxane continue unchecked in an environment in which prostacyclin is absent. The fact that the comparator drug in the other arm had the potential, if taken regularly, to block the formation of thromboxane to a very substantial extent was clearly a circumstance of real significance in the interpretation of the results of VIGOR. Dr Reicin and Merck were not, in my opinion, unreasonable in having so regarded it. I would also add that I have not had the benefit of Dr Reicin’s reaction to the suggestion that the presumptive antiplatelet effect of naproxen ought not to have been regarded by her and her colleagues, in 2000, as implying cardioprotective consequences. She was extensively cross‑examined about her perceptions at the time as to the legitimacy of the conclusion which she drew in March 2000 and thereafter that the interpretation of the VIGOR results which was most consistent with all the data, and with what was known about naproxen, was that naproxen had operated in a way which was cardioprotective. She was also cross‑examined with respect to her reliance on Protocol 061. She consistently used the word “cardioprotective” as a compendious reference to the clinical consequences of the antiplatelet action of naproxen. Her attention was not drawn to the distinction upon which the applicant now seeks to rely.
603 Finally with respect to this group of submissions on behalf of the applicant, it is true that aspirin was not a comparator in Protocol 061. However, the biochemical action of aspirin on COX-1 was a known entity in March 2000. It was known that aspirin irreversibly inhibited platelet COX-1. I consider that the scientists at MRL were reasonable to take the results of Protocol 061 for what they demonstrated at the pharmacological level with respect to naproxen standing alone. They demonstrated biological efficacy with respect to the inhibition of platelets over the dosing period, if naproxen were regularly taken twice daily. To draw this conclusion it was not, in my opinion, necessary to have aspirin as a comparator. Neither was it suggested to Dr Reicin in cross‑examination that the interpretation which she placed upon the Protocol 061 data was somehow compromised by the absence of aspirin as a comparator.
604 For the above reasons, I am not persuaded that it was unreasonable of Dr Reicin to have used the data from Protocol 061 in the way she did in attempting to interpret the results of the VIGOR trial. However, she did not do so in isolation. An important aspect of that interpretation was the data available to her from the other trials of Vioxx which had then been completed, or which were ongoing. The applicant had submissions to make along that axis also, and I shall turn to them presently.
605 The applicant was also critical of Dr Reicin’s reliance upon Brochier (1993) and Fornaro (1993) referred to in paras 82 and 83 above. With respect to the flurbiprofen study by Brochier, he relied upon the following evidence by Prof Zipes:
To further support her conclusion, … Dr. Reicin cites a study by Brochier on flurbiprofen ... an NSAID tested in patients after successful treatment for acute [myocardial infarction] by thrombolysis and/or coronary angioplasty.… Flurbiprofen showed prevention of reinfarction and reocclusion of the infarct related artery. Citing this study as proof of her argument of the benefits of naproxen to me is totally misleading. First, flurbiprofen may or may not be similar to naproxen. As Dr Reicin herself points out from study 061, all NSAIDS are not the same, and she provides no comparison of naproxen with flurbiprofen. Second, the population tested is post thrombolysis or angioplasty, and not at all similar to the rheumatoid arthritis population tested in VIGOR. Therefore this study cannot be used to support the alleged protective effect of naproxen.
Prof Celermajer expressed a different view. He noted that, because of ethical considerations, “the area of trials in arthritis does not lend itself easily to placebo‑controlled trials, as patients with arthritis require some medication for pain relief”. He said that, in the absence of an RCT comparing naproxen with placebo, “several lines of evidence” had been proposed to support the naproxen hypothesis, one of which was the data from Brochier (1993) and Fornaro (1993). As to the former, Prof Celermajer said:
In a trial by Brochier …, Flurbiprofen was associated with a marked decrease (approximately threefold) in the reinfarction rate and the halving of the need for coronary revascularisation in the six months following treatment of patients for an [acute myocardial infarction], after thrombolysis and/or coronary angioplasty. This is certainly a highly select group of patients, however the author concluded that Flurbiprofen appeared to be an effective drug for the prevention of reinfarction after coronary reperfusion, most probably via an anti‑platelet effect.
606 Profs Zipes and Celermajer were likewise at odds with respect to Dr Reicin’s reliance on Fornaro (1993). Prof Zipes said:
[Fornaro (1993)] was a test of indobufen to prevent clot development and emboli (travel of the clot in the bloodstream) in patients with atrial fibrillation who are at risk for such events. … [T]here is no evidence that indobufen is similar to naproxen. In addition, the population tested with indobufen had atrial fibrillation, an arrhythmia (fast heart beat from the top part of the heart, the atria) that predisposes to clot formation in the atria by a totally different mechanism than the clots formed in the coronary artery from coronary artery disease. Citing this article to support the alleged benefit of naproxen in VIGOR is misleading and inappropriate.
Prof Cerlermajer said:
Also in 1993, Fornaro et al published a study … suggesting that Indobufen might reduce the risk of ischaemic events in patients with heart disease associated with an increased risk of embolism, with an approximately threefold decrease in the primary endpoint. Again, the authors ascribed this effect to inhibition of platelets.
607 Neither Prof Zipes nor Prof Celermajer was examined as to his views on the relevance to the VIGOR results of the Brochier and Fornaro articles. Dr Reicin was, however, cross‑examined on the reasonableness of her reliance on the articles. It was put to her that only if it be assumed that naproxen and flurbiprofen were the same in their inhibition of platelet aggregation would the Brochier article have been of any value to her. She replied:
I didn’t think of it in that regard in that here was an NSAID and it was showing in a high risk patient population that it was acting as a cardioprotective agent. I knew of no other reason why an NSAID would act as a cardioprotective agent.
It was put to Dr Reicin that she had no information enabling her to know whether flurbiprofen was comparable to naproxen. She responded that the naproxen and flurbiprofen labels both referred to the prospect of bleeding time being increased for patients who took those drugs. She was asked whether the labels were the only thing that enabled her to form a view about whether flurbiprofen and naproxen were relevantly similar. Dr Reicin did not accept that, saying that the fact that there was a non‑selective NSAID that had a cardioprotective effect showed her that NSAIDs could be cardioprotective.
608 I do not know what Dr Reicin would have made of Prof Zipes’ evidence that neither Brochier (1993) nor Fornaro (1993) provided a clinically apposite precedent for the presumed cardioprotective effect of naproxen observed in VIGOR. She made no comment on Prof Zipes’ evidence in her statement in reply, and she was asked about it neither in chief nor under cross‑examination. Notwithstanding these omissions, I had the clear impression from Dr Reicin’s evidence that she and her colleagues at MRL did not rely upon these articles as conclusive of the problem which confronted them in 2000; nor did they assume that naproxen was necessarily the same as flurbiprofen and indobufen in relevant respects. Rather, they were individual pieces of scientific evidence that helped them put the VIGOR results in context. The approach which they took generally appears from the following answer given by Dr Reicin to the question “What additional [ie additional to Brochier (1993) and Protocol 061] evidence did you get after 9 March [2000] that satisfied you of the cardiovascular safety of Vioxx?”:
We went back to the [osteoarthritis] database and re‑analysed that data to ensure that in fact we had not missed a cardiovascular safety signal and in fact, once again saw that the rate of serious cardiovascular events was similar on Vioxx and the other NSAID comparators. So that was an important piece of information. The Alzheimer’s data was also quite important. When you looked at the incidence of serious cardiovascular events in that study the rate, if anything, was lower, numerically, on Vioxx than it was on placebo. So those were two very important pieces of information. So once you understood that Vioxx didn’t have a rate that was increased … compared with placebo in Alzheimer’s, didn’t have an increased rate compared to other NSAIDs but it had an increased rate in VIGOR compared to naproxen, so how do you put that all together? That’s when the clinical pharmacology that Barry Gertz showed me was very important, because it explained how naproxen was different than the other NSAIDs we had in [osteoarthritis] … and that naproxen’s level of inhibition of platelet aggregation in thromboxane was similar to what you would see with aspirin if you took it at a dose of 500 milligrams twice daily continuously. I also saw the flurbiprofen study which had evidence in a controlled clinical trial of an NSAID acting as a cardioprotective agent with the number of heart attacks in that study actually looking quite close to what we saw in the VIGOR study. There is another NSAID called indobufen that was actually on the market in Europe as a cardioprotective agent. ... We had gone through the [Worldwide Adverse Experiences] database to look for a cardiovascular signal. We did not see one. Dr Braunstein had gone through to look at all incidences of [adverse experiences] that were reported in patients out in the marketplace who had lupus who had taken the drug. There were no thrombotic events of concern that had been reported. So it was the totality of data that led me to the conclusion that Vioxx was not causing heart attacks.
609 The applicant then submitted that, even if Merck were justified in assuming that naproxen possessed cardioprotective properties, there was no basis for an assumption that it was as cardioprotective as low‑dose aspirin. However, this submission was not developed with any detailed attention to the evidence and, as I have attempted to explain elsewhere, the state of science now is that naproxen, taken regularly, can have an antiplatelet effect similar to that of low-dose aspirin. All the cardiologists gave evidence to that effect in their joint report.
610 The applicant next submitted that, in March 2000 and subsequently, Merck ought to have appreciated that, even if it be accepted that naproxen had the potential to be about as cardioprotective as aspirin, the size of the relative risk in the VIGOR data was simply too great to be explained by that circumstance. Here the applicant’s case dealt chiefly with myocardial infarction, in which respect I have dealt with the objective likelihood of the data being explained by the cardioprotective effect of naproxen at paras 371‑383 above. Dr Reicin was cross‑examined extensively on this. She accepted that the report submitted to the FDA on 23 March 2000 (of which she was the main author) did not make any attempt to grapple with the issue presented by this aspect of the VIGOR data. I would not criticise Dr Reicin, or Merck, for not having done so as at 23 March 2000, since the report to the FDA of 23 March 2000 was what was described as a “14‑day report” required by the FDA and the data available then were, to the extent of about 50%, based on unadjudicated events. However, do the subsequent, and more considered, reports to the FDA demonstrate that Merck addressed this issue?
611 The author of the analysis appended to the Clinical Study Report of 29 June 2000 identified the factors which, in some unquantified combination, might well have explained the VIGOR data. The cardioprotective effect of naproxen was one such explanation, but the author did not proceed to ask explicitly whether, if such an effect were in play, it went the full distance required to explain the apparent 76% reduction in risk of myocardial infarction associated with taking naproxen (as was implied by the relative risk figure of 0.24 (0.08, 0.70) referred to in the analysis). The analysis referred to the disproportionately large number of myocardial infarctions suffered by patients who, in violation of inclusion criteria, were aspirin‑indicated, but there seems to have been only a slight asymmetry in the distribution of these patients as between the arms (Vioxx 170; naproxen 151), and this circumstance was not offered as an explanation for the overall disparity of the results.
612 That disparity was sought to be explained by one or other, or a combination, of the naproxen effect and the atypical susceptibility of rheumatoid arthritis sufferers to CVT disease. As to the latter, Dr Reicin was cross‑examined as to when she first formed the view, referred to in her witness statement in reply (see para 377 above), that the systemically inflammatory condition of rheumatoid arthritis sufferers might have accentuated the cardioprotective effect of naproxen in the VIGOR trial, based as it was on Ridker (1997). She accepted that she had not come across Ridker when she wrote the preliminary report to the FDA on 23 March 2000, but she said that she had “referenced” the article in what was sent to the FDA in June. Although she was not challenged on that evidence, for my own part, I can find no relevant reference in the Clinical Study Report to Ridker (1997). The only references to an article by Ridker are to Ridker (1998) and to Ridker (2000). However, the essence of the point was mentioned in that report, in about the middle of the lengthy passage I have set out in para 129 above. The report there dealt with the possibility that rheumatoid arthritis sufferers might be such as would derive an exaggerated cardioprotective benefit from the ingestion of an antiplatelet agent such as naproxen was supposed to be. These few lines stand as the only evidence of Merck having held the possibility out as an explanation for VIGOR until the clear articulation of it in the FDA Advisory Committee Background Information package in 2005. As I read Bombardier (2000), there is no treatment of this explanation. The slides which Dr Reicin and Dr Nies showed to the FDA Arthritis Advisory Committee on 8 February 2001 do not refer to it (notwithstanding that they assert equivalence of naproxen to aspirin). Neither does the transcript of their oral presentation that day. In this proceeding, the matter was mentioned first in Dr Reicin’s witness statement in reply. Her original witness statement in this case, in which she (apparently exhaustively) set out every angle of Merck’s attempts to understand the VIGOR results, made no reference to it. In all the circumstances, I am disposed to accept the way it was (rather tentatively) put by the respondents in their final written submissions: “Dr Reicin and others at Merck hypothesised that naproxen’s protective effect could be magnified in the VIGOR population”.
613 I accept Prof Vaughan’s evidence that Ridker (1997), which he identified as the “physicians’ health study”, provided a basis for such a hypothesis. Additionally, there are two other circumstances which bear upon the question of the reasonableness of Merck’s response to the VIGOR results. The first is dose. I refer to my conclusion in para 463 above. The dose of Vioxx administered in VIGOR was 50 mg daily – twice the dose in APPROVe and twice the recommended dose in Australia over the period to which this proceeding relates. I consider that any reasonable interpretation of the VIGOR results should have allowed for some dose effect. The second is what Prof Celermajer referred to as the fragility of the relative risk figure where small numbers are concerned. This relates to the question which I left open in para 386 above. Indeed, given that VIGOR was a trial in which prophylactic aspirin was not permitted and patients requiring it should not have been included, if one excludes the eight myocardial infarctions suffered by the small number of patients who were enrolled in violation of that limitation, there were 12 events in the Vioxx arm and four events in the naproxen arm – in a trial involving over 8,000 patients. It was, in my view, not unreasonable of Merck to have baulked at concluding from these figures that Vioxx probably caused or contributed to myocardial infarctions generally.
614 All things taken into account, I do not consider that the VIGOR results, even standing alone, were such as required Merck, as a manufacturer with a duty of care, to draw such a conclusion; or to draw a similar conclusion in the case of CVT events generally. Indeed, I do not believe that the cardiologists (save possibly Prof Zipes) proposed that VIGOR itself required such a conclusion to be drawn. They expressed the view, which I accept, that VIGOR stood as a “potentially worrisome and potentially important signal of cardiovascular risk”. I also accept their opinion that this signal “mandated comprehensive analysis and investigation”. Although that was a scientific opinion, it happens to correspond with the view I hold as to the obligation which thereupon fell upon Merck pursuant to its duty to take reasonable steps to avoid harm being done to those to whom it owed a duty of care.
From VIGOR to 31 December 2000
615 The applicant submitted that the “vast majority” of Merck’s clinical trials provided evidence of increased CVT risk associated with the consumption of Vioxx. He relied upon Prof Madigan’s re‑analysis of the data available to Merck from its arthritis trials of Vioxx as at 31 December each year from 1997 to 2003 (see para 393 above). However, for reasons given at para 399 above, I am not persuaded that it was unreasonable of Merck not to have included the six studies which Prof Madigan added to those that provided the basis for Merck’s own analysis as forwarded to the FDA. Although Prof Madigan provided a single relative risk figure which was based on the combined rheumatoid arthritis and osteoarthritis data other than from those six studies (the figure to which I have referred at para 392 above), his year‑on‑year table was not so limited.
616 There is also a question whether it was reasonable for Merck, in the discharge of its duty of care, not to have separated out the combined rheumatoid arthritis and osteoarthritis data from the Alzheimer’s disease (and low back pain) data in the way that Prof Madigan did for the purposes of this litigation. In his witness statement, Prof Madigan referred to a January 1999 document of Merck’s titled “Analysis Strategy for the Incidence of Serious Thromboembolic Events in COX-2 Specific Inhibitor Program” which, he said, specifically called for a combined analysis of the data from the osteoarthritis and rheumatoid arthritis placebo‑controlled studies separate from that of the Alzheimer’s disease studies. No objection was taken to that evidence by Prof Madigan, but there was some ambiguity about the document of January 1999 to which he referred. A document (discovered by the respondents) dated 6 January 1999 with the same title as that mentioned by Prof Madigan was tendered by the applicant on the understanding that it was the document in question, and the respondents accepted that it was, but, in place of the spreadsheet which ought to have provided support for Prof Madigan’s evidence, there was a notation “Table 1 (… Spread Sheet goes here)”. According to the text of the document, Table 1 contained “details of ongoing and currently planned trials covered by the [CV] SOP”. Further confusion was added by the circumstance that Prof Madigan’s footnote for this document referred to the discovery number of a document in other litigation, and the document which was tendered did have such a number, but it was not the same number as that mentioned in the footnote. In the result, the applicant relied on Prof Madigan’s evidence on the basis that he was not challenged on it.
617 The respondents made their submissions on the footing that the tendered document most probably was the one which Prof Madigan had in mind. They pointed out that it was a draft, and that it stated: “A formal data analysis plan will be drafted in 1999 and approved before the first analysis is conducted.” I am disposed to accept those submissions. I think the coincidence of date and title is such as to make it most likely that this was the document to which Prof Madigan referred. I do not place much store by the anomaly in the discovery numbers. Further, even absent the apparently significant spreadsheet, it is possible to derive a fair understanding of what one author of the document had in mind. It was proposed that analyses of data from trials of coxibs be undertaken in three different “blocks”, one of which was expressed as follows:
VIGOR (RA),
VIOXX studies by MRL, CDP, and CDSP (RA and OA), and
VIOXX studies for 4Q01 WMA [RHEUMATOID ARTHRITIS]
The missing spreadsheet was a “see also” reference for this part of the document. (I understand that the initials “CDP” indicate “clinical development program” and that the initials “CDSP” indicate “clinical development studies program”.)
618 What is material for present purposes is whether Merck itself should be held to the proposal of the author of this document, in effect as an admission that it was appropriate to pool the arthritis data only. I do not think so. There was no evidence as to the identity of the author of the document or as to his or her place in Merck’s organisation. Self‑evidently the document is a draft of some kind, and should not be taken as representing the position of Merck as such in January 1999.
619 Was it nonetheless reasonable for Merck to have looked only at the arthritis data as part of a single pooled analysis in which the Alzheimer’s disease (and low back pain) data were also included? Save to point to the signal of toxicity that would have been revealed by the arthritis data alone, the applicant did not propose any reason why it was not. I have referred to Dr Reicin’s justification for Merck having looked at the data in the way that it did at para 395 above. I can see no reason not to accept that justification.
620 It was submitted on behalf of the applicant that, whether or not the arthritis data might have been pooled with the Alzheimer’s data, they ought also to have been looked at in their own right, if for no other reason than that they represented the cohort of patients most likely to be prescribed Vioxx. If specific clinical relevance is to be the touchstone, however, one might observe that rheumatoid arthritis and osteoarthritis are separate diseases, and the data relating to each of these entities, considered separately, did not reveal a statistically significant risk related to taking Vioxx rather than placebo. By pooling those separate data sets, Prof Madigan derived a combined figure that was statistically significant. If one takes those steps, however, it might be asked: why not improve the quality of the data even further by adding the Alzheimer’s results to the pool? Absent heterogeneity, I can think of no obvious answer to this question. The position in which Merck found itself after March 2000 required an understanding of the association between taking Vioxx and the occurrence of CVT events. The VIGOR trial had provided naproxen‑controlled data only. It was necessary to look for placebo‑controlled data. Had Merck looked at the arthritis data alone, it might well (at various times after March 2000) have seen a statistically significant signal of risk. However, had it also looked at the Alzheimer’s data alone, it would have seen a non‑significant signal that Vioxx was cardioprotective. In these circumstances, it was entitled to take the view, which it did, that the pooling of all available placebo‑controlled data was the surest way of understanding the complete picture. It may not have been the approach which another manufacturer in the position of Merck might have taken, but it was the approach which Merck in fact took and I am not persuaded that it was unreasonable for it to have done so.
621 The applicant next turned to the Alzheimer’s studies as such, contending that, properly understood, the data from them put Merck on notice that the consumption of Vioxx was likely to have increased the risk of CVT events, of death and of CVT death. Here he relied upon various relative risk calculations undertaken by Prof Madigan with respect to data that were available to Merck at different points in time. This was done in three ways. The first was done by reference to studies which had concluded as at 31 December in the year in question. Here Prof Madigan’s relative risk figure was 1.0 (0.5, 2.1) as at 31 December 2000. The second included data from all studies, even those which, at the date in question, were ongoing. Here the figure as at the same date was 1.3 (0.09, 1.9). The third set out the dates upon which the risk of taking Vioxx became “permanently statistically significant”. Here it was only with respect to mortality that there was a statistically significant excess in the Vioxx arms of the studies by the end of 2000. Using investigator‑reported data, the excess was significant on 15 January 2000 (with a relative risk of 6.5). Using the data for confirmed CVT deaths, the excess was significant, for Protocols 078 and 091 combined, on 28 November 20000 (with a relative risk of 3.4).
622 Save with respect to “confirmed CVT Deaths”, Prof Madigan’s analyses were based upon investigator‑reported events. For the reasons given above at para 410, I would not hold that it was unreasonable for Merck to have preferred, as it did, confirmed events as a basis for the discernment of risk associated with the consumption of Vioxx. I accept the respondents’ case that, in a matter as serious as that which was the subject of the CV SOP, to have relied only on the categorisation of events given by investigators, and not to have made use of specialist, blinded, adjudicators would rightly have been regarded as sub-optimal.
623 Prof Madigan’s CVT mortality figures were based on deaths whenever they occurred. But I do not hold it to have been unreasonable for Merck to have collected and analysed CVT events by reference to its “modified ITT” approach, ie one which involved counting only the events which occurred on‑drug or within 14 days after discontinuation. That approach was specified in the CV SOP which, as I have said, was developed by the medical scientists at MRL in 1998‑99 as a result of the advice they received from Merck’s Board of Scientific Advisers. It was not suggested by the applicant – either in his submissions or in cross‑examination of Dr Reicin – that the CV SOP was deficient in relevant respects. In her evidence set out at para 413 above, Dr Reicin provided a justification for the 14‑day approach which I cannot reject as unreasonable. The highest the applicant’s experts put their criticism of Merck in relevant respects was that, had Merck looked beyond its 14‑day data to the data that would have been yielded by a full ITT study, it would have perceived a stronger signal of risk than it did. That, however, is advice informed by the wisdom of hindsight. As Dr Reicin said, at the time when the CV SOP was being developed, the hypothesis which was presented to Merck proposed an acute, rather than a long‑term, effect. There was, she said, a view that to extend the collection of data well beyond discontinuation might cloud any signal of toxicity. The evidence in this case is not such as would permit me to hold that this view was not, as a matter of responsible and prudent medical research, reasonably open in 1998 and subsequently. It informed the content of the CV SOP. It was, in my view, reasonable for Merck to have applied the CV SOP to its Alzheimer’s studies.
624 I would say the same thing about the CVT mortality data specifically. Here it must be remembered that Merck counted deaths which occurred at any time, so long as they arose from adverse experiences occurring or commencing, either while the patients concerned were taking their drug (or placebo) or within 14 days after discontinuation. Thus the deaths counted by Prof Madigan but not by Merck were those which arose from an adverse experience which occurred more than 14 days after discontinuation. It was, in my view, reasonable for Merck to have applied the CV SOP in the way that it did. It should also be remembered that the generality of the Alzheimer’s data included both fatal and non‑fatal adverse experiences. It was not as though the deaths, which appeared to show a signal of toxicity which was missing from the general data, were not also part of that data.
625 Before leaving the Alzheimer’s mortality data as such, if one is to focus on data of that kind, one ought to look also at the like data generated by other studies and trials from which results were available to Merck in 2000. Dr Reicin said that those data did not throw up a signal of cardiovascular risk. Most conspicuously, perhaps, in the VIGOR trial itself there were the same number of cardiovascular deaths in each arm, yielding a relative risk figure of 1.00 (0.35, 2.86). For fairly obvious reasons, the applicant did not refer to that figure in his submissions. If the question presumptively occupying the minds of the scientists at MRL in 2000 was whether the consumption of Vioxx involved the risk of CVT death, I cannot understand why they would not have been entitled to take the view that the VIGOR results suggested not.
626 Of course, the Alzheimer’s data were important for Dr Reicin and her colleagues in March 2000 and subsequently more for what they did not show than for what they showed. As collected and reviewed by MRL, they showed (at least at that time) no signal that the consumption of Vioxx might be associated with cardiovascular risk. But the applicant advanced a number of respects in which the Alzheimer’s studies were characterised by “numerous problems in study design which obscured the risks”. The first was the apparent unsatisfactory level of compliance. I have dealt with that in paras 420‑422 above.
627 The second was the absence of a data and safety monitoring board. Here the applicant relied on the evidence of Prof Zipes that such a board was “the conscience for the trial performance”. Prof Zipes expressed this opinion generally, however, rather than specifically in connection with the Alzheimer’s studies. While under cross‑examination, Dr Reicin had put to her an MRL document entitled “Medical Affairs Procedures and Policies”. An appendix to Procedure 23 therein set out guidelines for data and safety monitoring boards. The appendix stated:
In order to ensure the safety of the trial participants it is often necessary to monitor the results of an ongoing trial in an unblinded fashion, and this monitoring is accomplished by a committee known as a Data & Safety Monitoring Board (DSMB).
And:
Most clinical trials done by Merck do not require a DSMB. One consideration is the seriousness of the medical condition under evaluation, and, therefore, the cost to the patient of receiving a less effective therapy. By identifying a drug whose efficacy exceeds expectations the DSMB can minimize the number of such patients. A second consideration is the level of uncertainty in the safety and efficacy of the experimental therapy. Patients are at much higher risk of unexpected serious side effects in the first large‑scale clinical trial of a new clinical entity, or in a trial involving a new patient population, than in a trial involving only marketed drugs in an existing patient population. Finally, the need for a DSMB is stronger in trials involving a large number of participants and in lengthy trials than in short trials involving few subjects.
Dr Reicin agreed that Alzheimer’s disease was a serious medical condition, but added that there were not good therapies for the disease “so putting them on placebo versus Vioxx wouldn’t have put the patients at risk of being on a less effective therapy.” She agreed that Protocol 078 was a long trial. From these considerations, and from the number of patients in the trial, counsel for the applicant put it to Dr Reicin that Protocol 078 was such as required a board. She had not been involved in the decision not to have a board (or, more accurately, to dispense with the board that had originally been specified), and could not comment.
628 In their written submissions, counsel for the applicant contended that “Merck’s failure to include a DSMB in the Alzheimer’s studies eliminated safeguards and introduced bias, weakening the reliability of the study.” That proposition was not put to Dr Reicin. Neither, so far as I could tell, did it find expression in the evidence of any other witness (none was referred to in the written submissions). It is not a self‑evident proposition. In the context of a study in which all reported CVT events are to be sent for adjudication, it is not at all apparent to me how the absence of a board whose members were unblinded to the data would “introduce bias” or “weaken the reliability” of the cardiovascular data collected. Specifically, so far as the applicant’s case is concerned, I was not favoured with any explanation as to how the absence of a board ought to have been regarded by Merck as compromising its ability to take the results of the Alzheimer’s studies at face value.
629 The third respect in which it was submitted that the Alzheimer’s data were disqualified from use in the way that Merck used them was that the studies “were not designed or powered to detect CVT events”. No evidence was referred to in support of this proposition. It is true, however, that the studies were not “designed” for that purpose, but then neither were the arthritis studies (and neither, for that matter, was VIGOR). That is why Merck instituted the CV SOP. That is also why Merck pooled the Alzheimer’s data with the like data from other trials and studies.
630 The fourth such respect was that “the inability of dementia patients to self‑report limited proper reporting of adverse events, further underpowering the study – and more patients converted to Alzheimer’s disease on VIOXX, further skewing the results in favour of VIOXX”. In support of this submission, counsel referred to the report submitted by MRL to the FDA on 24 January 2005. Dealing with Protocol 078 (prevention of Alzheimer’s disease), the report did note that there were more conversions to the disease in the Vioxx arm. However, the first limb of the proposition set out in the extract from counsel’s submissions finds no support in the report – definitely not at the references given, nor, so far as I can see, anywhere else.
631 The fifth respect was that the trials “excluded patients with major stroke, multiple lacunar infarcts, or transient ischaemic events within two years, [myocardial infarction], CABG, angioplasty, or stent placement within one year, a history of angina or congestive heart failure, or uncontrolled hypertension”. These circumstances were, in each of the relevant protocols, amongst a lengthy list of exclusion criteria. That they were inappropriately so was not put to Dr Reicin. Similar exclusion criteria were specified in the VIGOR and APPROVe trials, upon the results of which the applicant relies. I consider that there is nothing in this point.
632 The final respect in which, the applicant submitted, the Alzheimer’s studies had problems of design which obscured the CVT risk signal was that the subjects were permitted to take low‑dose aspirin. That was so: patients who, after randomisation, developed a need to take low‑dose aspirin for cardioprotective purposes were permitted to do so. Intuitively, I can understand why, in the context of the FitzGerald hypothesis, such a dispensation might well present the potential to compromise the utility of the Alzheimer’s disease data for cardiovascular safety purposes. However, in their submissions, counsel took the matter no further than to make the fairly bald assertion to which I have referred. The point was not the subject of any evidence led by the applicant. Neither was it put to Dr Reicin, or to any of the respondents’ experts under cross‑examination. Indeed, when the court suggested (in the course of a more general exchange) to Dr Reicin that she might not have expected to see the result predicted by the FitzGerald hypothesis, even if that hypothesis were valid, in the Alzheimer’s studies because the patients in those studies were permitted to take low‑dose aspirin, Dr Reicin replied:
It was permitted in the Alzheimer’s study but very few patients took it and we did do sensitivity analyses and even if you took those patients out, the event rate was similar between Vioxx and placebo.
In the circumstances, I am not prepared to hold that the utility of the Alzheimer’s data should have been regarded as of limited utility because of the ability of patients to take low‑dose aspirin.
633 Thus I would hold that it was not unreasonable for Merck to have informed itself of the potential for Vioxx to increase the risk of CVT events by reference to the Alzheimer’s data as collected and as reported to the FDA. Those data conveyed no signal of risk. Indeed, as Merck pointed out in some of its reports to the FDA, the patient groups in the Alzheimer’s study tended to be the elderly, amongst whom a signal of CVT risk might readily become apparent if Vioxx were indeed having a harmful effect.
634 For the foregoing reasons, I am not of the view that the respondents ought to have known by the end of 2000 that Vioxx caused or contributed to myocardial infarctions or to CVT events generally. The position remained as it had stood on the release of the VIGOR results, namely, one in which the respondents were confronted with a worrisome signal of potential cardiovascular risk.
2001
635 A number of things happened in 2001 with respect to the emerging picture of the association between the consumption of Vioxx and cardiovascular disease. Two pooled analyses were forwarded to the FDA – one on 8 January and the other on 12 July. There was a meeting (partially in public) of the FDA Arthritis Advisory Committee on 8 February. The final results of Alzheimer’s Protocol 091 (which had ended on 30 November 2000) and those of Protocol 126 (which was terminated early on 31 March) became available for analysis. Konstam (2001) and Reicin (2002) were submitted for publication. A warning letter was received from the FDA. And there was much discussion within MRL as to the need for, and the possible architecture of, a cardiovascular outcomes study. I shall commence with the Alzheimer’s data.
636 In the final data from Protocol 091, the relative risk of encountering a CVT event from taking Vioxx rather than placebo was shown to be 0.39 (0.12, 1.21). The like figure in the case of Protocol 126 was 1.25 (0.38, 4.10). The wide confidence intervals involved in these figures demonstrate the utility of a pooled analysis. The figures did, of course, make their contribution to the pooled analyses forwarded to the FDA by Merck, specifically (in the case of these final data) to the third such analysis dated 22 May 2002. There was nothing in them which indicated CVT risk.
637 It was submitted on behalf of the applicant that the “all‑cause mortality” data available from the Alzheimer’s trials as at April 2001 showed a clear excess of events in the Vioxx arms, and ought to have been recognised by Merck as a signal of cardio‑toxicity. This relates to the subject of Dr Reicin’s evidence set out at para 448 above. On 8 April 2001, Dr Joshua Chen, a statistician in MRL, prepared a memorandum analysing the mortality figures yielded by Protocols 078 (which was ongoing) and 091. He took two approaches – ITT and on‑drug. Combining the data from the two protocols, Dr Chen calculated that the relative risk of death, from taking Vioxx rather than placebo, was 2.99 (1.55, 5.77) taking the ITT approach and 2.68 (1.23, 5.82) taking the on‑drug approach. These were only two of several ways in which Dr Chen analysed the data, but, on any view, there appeared to be a statistical association between the consumption of Vioxx and death from any cause. The one analysis which did not produce a statistically significant result favouring placebo was the on‑drug analysis of the data from Protocol 078 standing alone, where the relative risk of taking Vioxx was 2.07 (0.82, 5.19).
638 As mentioned in para 448 above, Dr Reicin said that cardiovascular mortality was not the “key driver” behind these figures. Merck looked at the actual causes of the deaths that had produced an excess in the Vioxx arms and, according to Dr Reicin, found that many of them were manifestly not related to treatment. The legitimacy of taking this kind of approach to statistics was debated in the joint cardiology session by reference not to the data available to Merck in the Spring of 2001 but to the FDA review of 18 December 2004. Prof Zipes said that the whole purpose of an ITT approach was to record all adverse events occurring during the period of the study, but accepted what was put to him by counsel for the respondents, namely:
[W]hilst the all cause mortality approach may throw up some numbers that are required to be looked at, they need some careful examination and analysis to see if deaths such as a car accident or electric shock can be related in some way or other to the drug under study.
Prof Harper said:
I think the principle of intention to treat is very important because you could have a heart attack and have a car accident as a result of it. You could suffer death from an electric shock because your heart was weaker than it otherwise would be. So if you have a truly randomised study then those factors should come out in both arms so I think it’s important to restate the importance of the intention to treat principle.
Prof Celermajer agreed with Profs Zipes and Harper, but noted (of the 2004 data) that “infections and inflammations went 6 nil against rofecoxib and on the surface of it, it’s hard to attribute that to rofecoxib”. He added that it was important to “[look] at each of these carefully”.
639 I consider that the approach taken by Merck to the all‑cause mortality data in the Alzheimer’s studies in 2001, and thenceforth until Vioxx was withdrawn from the market, was a reasonable one. Scientists at MRL recognised the excess of deaths in the Vioxx arms of the studies, but, because of the manageable numbers involved, were able to drill down and to obtain clinical – ie not merely statistical – information about those deaths. This was clearly a prudent and responsible approach, calculated to provide a more satisfactory basis for any decisions that had to be made.
640 The same response is not available in relation to the confirmed CVT deaths arising from the Alzheimer’s studies. In late May 2001 a draft of the report which was ultimately sent to the FDA on 12 July 2001 (containing the second pooled analysis) showed the deaths – both total and confirmed CVT – in each of the arms of Protocols 078, 091 and 126. The text of the draft had been prepared on the basis of showing the deaths which occurred at any time during the conduct of the studies. Dr Reicin amended the text by limiting it to deaths (whenever they occurred) which arose from adverse events occurring up to the 14th day after the last dose of study medication. Dr Reicin defended these amendments by noting that they were made only in the text of the report, adding:
[T]he appendix to the final version of that Safety Update Report contains adjudication packages for all the deaths in the Alzheimer’s studies up to that point, and so information about the off‑drug deaths was in fact given to the FDA in that document. I believe that this approach was entirely appropriate.
I set out the effect of Dr Reicin’s changes in the table below (where the changed figures are showed in parentheses; note also that some of the figures in the “CVT” column for Protocol 078 include events described as “sudden unexplained death”).
|
Total Deaths |
CVT Deaths |
|||||||
|
Vioxx |
Placebo |
Vioxx |
Placebo |
|||||
|
078 |
22 |
(15) |
10 |
( 9) |
8 |
( 5) |
2 |
(2) |
|
091 |
16 |
(14) |
9 |
( 8) |
3 |
( 3) |
2 |
(2) |
|
126 |
4 |
( 4) |
4 |
( 3) |
1 |
( 2) |
1 |
(1) |
|
All |
42 |
(33) |
23 |
(20) |
12 |
(10) |
5 |
(5) |
641 The applicant made much of Dr Reicin’s intervention to amend the figures which ultimately found their way into the report of 12 July 2001. The implicit charge against her (which was not, as it happens, put to her squarely) was that she and her colleagues at MRL were massaging the figures that would find public expression in the report to the FDA in order to minimise the appearance of cardiovascular toxicity. There would be no substance in such a charge. As I have said above, the CV SOP required the counting of events which occurred before the 14th day after discontinuation. It was appropriate, in my view, that Merck apply this guideline by counting deaths which arose at any time from adverse experiences which occurred within that period. As it happens, Dr Reicin’s intervention made very little difference to the figures: overall, the deaths in the Vioxx arms were reduced from 12 to 10 and those in the placebo arms were unchanged at 5. It might also be noted that the effect of Dr Reicin’s intervention was not uniformly to cast Vioxx in a more favourable light than was the case under the figures in the original draft. An aspect of her amendments to the draft was, it seems, to require a more careful look to be had at the categorisation of deaths which had not been recorded as CVT‑related, the result of which was to increase by one the number of deaths in the Vioxx arm of Protocol 126 which were so related.
642 However these figures are looked at, they ought, standing alone, have been seen as a conspicuous signal of CVT risk. How were they in fact seen by Merck? The Safety Update Report of July 2001 provided the following commentary:
Compared with the placebo group, the rofecoxib group had 5 more deaths adjudicated as due to thromboembolic cardiovascular events (10 versus 5). Review of the thromboembolic events shows a variety of events, including 2 patients in the rofecoxib group who were also taking aspirin and had intracranial bleeding (Protocol 126 ANs 532, 743). The proportion of patients with a serious cardiovascular adverse experience (fatal or nonfatal) was similar in the 2 groups (rofecoxib, 5.3%; placebo, 5.7%), suggesting the difference in deaths was not due to a pharmacologic effect. This is supported by the analysis of confirmed thromboembolic events in the Alzheimer Studies … and the cardiovascular meta‑analysis of data from 23 clinical trials …
This suggests to me that Merck recognised the excess of CVT deaths in the Vioxx arms, placed it in the context of all the data that were available, and sought to understand its potential significance to the question of cardiovascular risk associated with Vioxx.
643 The applicant relied on what was said to be an internal Merck document, discovered by Merck and tendered by the applicant, but not put to or explained by any witness. It has the appearance of a series of slides, and is headed “Outline of Presentation”. It is not dated, but, since it states that Protocol 091 had been completed and that Protocol 126 had been terminated, but makes no similar comment about Protocol 078, I infer that it was prepared subsequently to March 2001 but before April 2003. The applicant made no attempt to correlate the figures in this document with any others before the court, or to give the document a date or context generally. He relied merely upon a comment in the “Summary” section of the document that “[i]mbalance in death in Alzheimer studies was driven by an imbalance in [cardiovascular] death, respiratory death (mainly pneumonia), and trauma”. However, the complete “Summary” was as follows:
- Imbalance in Death in Alzheimer Studies was driven by an imbalance in [cardiovascular] death, Respiratory Death (mainly pneumonia), and Trauma
- Number of events in each category are low
- Preliminary Data does not suggest a corresponding increase in [cardiovascular serious adverse events] or Pneumonia [serious adverse events] which might be expected if this was a “drug” effect
- Imbalance in Death was not seen in the Phase IIb/III [osteoarthritis] Program or VIGOR
Assuming that this was an internal Merck document prepared in mid‑2001 or thereafter, its terms, in my view, bespeak an awareness of the CVT mortality data generated by the Alzheimer’s studies, and a conscientious concern to put them in the context of similar data obtained from other trials and studies.
644 Turning from the Alzheimer’s mortality data to the pooled analysis as such, the situation in 2001 was not such as would warrant the conclusion on the part of Merck that the consumption of Vioxx gave rise to an increased risk of CVT disease. Only where the comparator was naproxen was there a statistically significant relative risk. Where the comparator was the generality of other NSAIDs, and where it was placebo, not only was there no such risk shown, but the non‑significant point estimate of relative risk fell below unity in both cases. It is true that, in each of these analyses, the data were (in the case of the second, primarily) presented in terms of the APTC endpoint, but, as was noted in the text of the second analysis, the incidence of haemorrhagic strokes was low (in fact there was a slight excess amongst those taking Vioxx), and exclusion of them would not have altered the conclusion. It should also be noted that the second pooled analysis set out the placebo‑controlled relative risk figure in relation to CVT events in the Alzheimer’s disease studies, and that was almost the same (the point estimate was the same) as for the APTC endpoint.
645 Since I have declined to accept the various bases upon which the applicant attacked these data, I would find it reasonable for Merck to have had recourse to them for such assistance as they provided. They were not the whole of the information then available, but they were available and they conveyed no signal of risk.
646 As will be apparent from what I have written at para 156 above, the outcome of the Arthritis Advisory Committee meeting on 8 February 2001 was not such as would have presented Merck with an expert consensus that the consumption of Vioxx gave rise to a risk of cardiovascular disease. By contrast, the consensus was that the prescriber and the consumer should be made aware that there had been an excess of CVT events in the Vioxx arm of VIGOR, and that the explanation for that was then unknown. That was not all good news for Merck, of course, as the possibility of the excess being due to the harmful effect of Vioxx was not excluded. Indeed, I am disposed to think that the position reached by the committee was similar to that expressed by the cardiologists in the present case, namely, that the VIGOR results stood as a “worrisome” and “important” signal of potential cardiovascular risk that required analysis and investigation.
647 Turning to the published research, the applicant was most critical of some aspects of Konstam (2001). He relied upon the report of Prof Woodward, who pointed out that 77% of the statistical weight of the pooled analysis was contributed by the data from the Alzheimer’s studies. He continued:
The fact that these trials were the only ones in the meta‑analyses that used incomplete, interim data is somewhat worrying and should have been picked out as an issue by the authors. My calculations suggest that pooling [osteoarthritis] and [rheumatoid arthritis] results alone produces a [relative risk] … of 1.59 (0.54‑4.72), suggesting a nonsignificant excess risk of rofecoxib, … This casts doubt on whether the three indication groups should, in fact, be pooled, although the low level of information does not allow for an accurate statistical test of heterogeneity.
Prof Woodward undertook a technical analysis of the data on which the Konstam article was based, and concluded that the pooled analysis was underpowered by conventional criteria. The authors, in his view, were not justified in saying that event rates in the two arms were similar, or were comparable. In their joint report, the biostatisticians agreed with Prof Woodward’s treatment of power in the context of Konstam, adding that the pooled analyses were sufficiently powered to detect “approximately a two‑fold or greater increased APTC risk and a 2.5‑fold or greater increased risk of [myocardial infarction]”.
648 The latter aspect of the first pooled analysis was not discussed in the Merck submission to the FDA, or in the Konstam article. Indeed, there is no suggestion that MRL saw the pooled analyses data in such terms at all. However, the opinion of the biostatisticians does show that the pooled analyses were sufficiently powered to detect a relative risk of an APTC endpoint of the general order of that observed in VIGOR, and a relative risk of myocardial infarction much less than that observed in VIGOR. Put differently, as I understand it, if the true relative risk of myocardial infarction (compared to placebo, point estimate) were 2.5 or greater, then these data would have shown it at a statistically significant level. While I accept, therefore, Prof Woodward’s evidence that the data did not justify a conclusion that there was no cardiovascular risk associated with taking Vioxx, at least they did not show the contrary and were, for all their limitations, quite obviously inconsistent with the VIGOR data. I am not presently concerned with the question whether Konstam and colleagues expressed the matter too sanguinely in their article: the present question is rather whether the position which Merck (qua manufacturer) adopted in the face of these data and all other relevant information was a reasonable one. Prof Woodward’s treatment of the article itself does not require a negative answer to that question.
649 Prof Woodward also criticised the authors of Reicin (2002) for their conclusion that, in analysing the data from Merck Phase IIb/III osteoarthritis trials, they “found no difference between rofecoxib, comparator nonselective NSAIDs and placebo” in the risks of CVT events (see para 171 above). He said that a more accurate statement would have been “found no evidence of a difference”, since the lack of statistical significance could mean either that there was in fact no difference or that there was insufficient evidence to detect a difference. Dr Reicin was cross‑examined extensively about this distinction which, ultimately, I think she regarded as little more than a matter of semantics. It is, however, an important distinction. Prof Woodward was not challenged on his treatment of the subject, which I accept. I also accept that Dr Reicin and her colleagues expressed the matter rather tendentiously in their article when they said that they had “found no difference”, since they had not scientifically found a non‑difference. Notwithstanding these reservations, the data were set out in the article for the reader to see and, such as they were, they did not convey a signal of cardiovascular risk. Merck would have been criticised, in the post‑VIGOR context, for not looking at them. It is true that the numbers were small, and that the capacity to draw meaningful conclusions was limited, but, as I understand it, it was for that very reason that these data were pooled with those from other trials in the pooled analyses that were sent to the FDA.
650 As to the distinction between “found no difference” and “found no evidence of a difference”, whatever may have been expressed in Reicin (2002), MRL itself viewed the totality of the data then available to it in the latter sense. Although the second pooled analysis, which was sent to the FDA in July 2001, contained some statements in the discussion section which were, perhaps, inappropriately categorical (as to which I refer to the extracts from the report which I have set out in para 167 above), that section also contained the following:
Among the non‑naproxen NSAID comparative studies there were 29 rofecoxib patients with confirmed APTC endpoint events (rate of 1.10 events per 100 patient‑years). In the non‑naproxen NSAID group, there were 17 patients with events (rate of 1.30 events per 100 patient‑years). Thus, there was no evidence of any difference for rofecoxib versus non‑naproxen NSAIDs in the risk of an APTC event.
And the following:
In the placebo‑controlled trials, there were 38 rofecoxib patients with confirmed APTC events during 2518 years of follow‑up for a rate of 1.51 events per 100 patient‑years. There were 34 patients with events in the placebo group during 2099 years of follow‑up for a rate of 1.62. The ratio of rates (95% CI) was 0.93 (0.57, 1.53). There is no evidence to suggest that rofecoxib has a clinically apparent prothrombotic effect.
651 At this point I should also mention an aspect of Konstam (2001) to which I have referred at paras 159‑161 above. In the course of the preparation of a draft of the article, Dr Morrison in effect questioned the appropriateness of pooling the Alzheimer’s data with those from the arthritis studies. The exchange of correspondence in August 2001 made it apparent that Dr Bolognese and the other statisticians at Merck were alive to the prospect that the data at their disposal might be diced and rearranged in different ways, or viewed through different prisms. The correspondence between Dr Morrison, Dr Bolognese and others in mid‑August 2001 demonstrates that they were. However, that correspondence also demonstrates, in my view, that those scientists took a conscientious and prudent approach to discerning just what it was that the data showed. I also consider that there is a distinction between actually or constructively knowing something, on the one hand, and using that thing as the guiding source of information for the purpose of drawing overall conclusions, and of taking action, on the other hand. At the latter level, I consider it not unreasonable for Merck to have instructed itself by reference to all the data, including that yielded by the Alzheimer’s disease trials.
652 The applicant relied on Mukherjee (2001), in support of the proposition that, in 2001, Merck ought to have recognised that Vioxx involved an increased risk of CVT events. Recognising that the CVT results of VIGOR might well have arisen from the cardiovascular benefits of naproxen, the authors examined the absolute myocardial infarction rates in that trial, and in a limited number of other Vioxx and celecoxib trials, with a view to comparing them with the like data in the placebo arm of a very large meta‑analysis of trials testing the cardioprotective effect of aspirin. They found that the Vioxx rate was 0.74 while the placebo rate was 0.52. It must be said that the authors themselves conspicuously stressed the limitations of their study, but it received fairly comprehensive criticism at the hands of the experts in the present case. Prof Celermajer said:
This study, however, used several historical control groups rather than contemporaneous groups from databases or controlled clinical trials. The subjects in VIGOR (Rofecoxib and Naproxen‑treated) all had [rheumatoid arthritis], whereas the historical controls did not. As [rheumatoid arthritis] itself is associated with an elevated risk of [cardiovascular] events … it could well be that the disease itself … was responsible for the elevated event rates compared with the control subjects reported by Mukherjee et al, rather than the effect of COX-2 inhibitor treatment. … Therefore, at best, these data should be regarded as hypothesis‑generating, because …, the use of historical controls is usually rather unreliable, especially compared to more acceptable methodologies.
Prof Woodward was rather less guarded:
This was a systematic review of clinical trials of COX-2 inhibitors which extracted only published data. This only identified four trials with published data on the association between COX-2 inhibitors and [cardiovascular disease] events, of which only three involved rofecoxib … Since this omits most of the trials, and the methods of analysis are either simplistic or highly dubious, this study adds nothing to my understanding of the body of evidence at 2001, but it does illustrate the paucity of data that was readily available to the general public and health professionals at that time. Furthermore, this work is important in that it highlighted the cardiovascular issues concerning rofecoxib, showing lack of consensus with the representations made by the authors and sponsors of VIGOR.
In other words, at best Mukherjee (2001) took the matter no further.
653 The applicant also relied upon the warning letter received from the FDA on 17 September 2001. It was submitted that this letter was part of the facts which demonstrated that Merck knew of the cardiovascular risk associated with Vioxx. However, the letter told Merck nothing that it did not already know. To the extent that the letter dealt with the science of the matter, it relied substantially on VIGOR. The letter was a complaint about Merck’s promotional statements in the USA, and does not advance an understanding of the matters presently under consideration.
654 The applicant also submitted that the ADVANTAGE trial, to which I have referred at para 107 above, gave a clear signal of cardiovascular risk. Although the trial was completed in 2000, it was in 2001 that most of the events to which I shall refer occurred. It seems that Merck had its moments with the FDA in relation to this trial. The Clinical Study Report (or preliminary or other report) for the trial was not in evidence (or if it was, it was not drawn to my attention), but the applicant relied upon an exchange of emails involving Dr Scolnick in early April 2001. It seems that the FDA had requested further information about advantage. The subject of the emails was “advantage cv events tables”. By the first email, Robert E Silvermann sent two tables to Dr Scolnick, with a copy to several people including Dr Reicin and Dr Douglas Greene. By the second email, Dr Greene informed Dr Scolnick that the FDA “may be trying to make something out of the 5 vs 1, 3 vs 1 and 3 vs 0 cardiovascular events in the aspirin‑taking subjects, even though the number of events is too small to interpret”. By the third email (to Dr Greene only), Dr Scolnick said:
Doug That is why asked for the tables. They have no data in the CDP study that is worth anything. Their action is heonous [sic] from my perspective. There are many actions they might have taken that would have been not to our liking but rational. This course is just stupid.
By the way over the years the reason we have resisted doing large marketing clinical studies is just this. It opens a lot of data to FDA that compromises the large clinically meaningful trials. Small marketing studies which are intellectually redundant are extremely dangerous and the PAC system with the marketing emphasis in CDP on all their studies opens pandora’s box which we have urged against from the beginning of time. … Now it turns out it has compromised the gorgeous Vigor study, the labelling we had wanted, and put us in a terrible situation/Ed
655 The scientists who conducted the advantage trial published their research as Lisse (2003). They said:
The rofecoxib and naproxen groups did not differ significantly in the number of thrombotic cardiovascular events, as defined by the combined Antiplatelet Trialists’ Collaboration end points (10 [0.4%] vs. 7 [0.3%]; P > 0.2) …, or in adjudicated confirmed thrombotic events (9 [0.3%] vs. 12 [0.4%]; P > 0.2). Five myocardial infarctions occurred in the rofecoxib group, and 1 occurred in the naproxen group (P > 0.2). No strokes occurred in the rofecoxib group and 6, all thrombotic, occurred in the naproxen group (P = 0.015).
Prof Zipes pointed out that the 5/1 myocardial infarction ratio seen in advantage was the same as, and therefore was consistent with, the like ratio in VIGOR; and there were 15 patients in the Vioxx arm who discontinued on account of hypertension, and only six in the naproxen arm who did so. Prof Zipes also went to a review of the advantage data written in December 2001 by two medical officers within the FDA. In a passage cited by Prof Zipes, they said:
Consistent with VIGOR, there was a trend of excess in serious cardiac thrombotic events in the rofecoxib 25 mg group, compared to the naproxen group (ten and three events, respectively, as per FDA review). There were five myocardial infarction …, two anginal events and three sudden deaths in the rofecoxib 25 mg group and one [myocardial infarction] and two angina (no sudden deaths) in the naproxen group. There were also two and five ischemic cerebrovascular events in the rofecoxib and naproxen groups, respectively.
656 The FDA reviewers were clearly dissatisfied with the adjudication process undertaken in relation to advantage. Because this aspect was not mentioned by the parties and neither Prof Zipes nor Dr Reicin was cross‑examined about it, I shall not deal with the causes of their dissatisfaction. However, they “re‑adjudicated” the data and presented the results in the following table:
Table 16. ADVANTAGE: Summary CV Thrombotic events as presented by the
sponsor (adjudicated by CV Adjudication Committee) and FDA re‑adjudicated events.
|
Number of patients with CV Committee adjudicated serious CV‑thrombotic events |
Number of patients with FDA re‑adjudicated serious CV‑thrombotic events |
|||
|
Rofecoxib (N=2785) |
Naproxen (N=2772) |
Rofecoxib (N=2785) |
Naproxen (N=2772) |
|
|
9 |
12 |
12 |
9 |
|
|
Cardiac Sudden death MI Angina Cerebrovascular CVA TIA Peripheral DVT/thromboflebit |
8 2 5 1 1 0 1 0 0 |
3 0 1 2 7 6 1 2 2 |
10 3 5 2 2 1 1 0 0 |
3 0 1 2 5 4 1 1 1 |
657 In her statement in reply, Dr Reicin referred to Prof Zipes’ opinion that advantage provided an important signal of cardiovascular risk, and said that it was “not supported by the data”. She referred to the passage from Lisse (2003) which I have set out above. Quite clearly, the data referred to in the Lisse article and the FDA data referred to by Prof Zipes cannot both be right. In their submissions, the respondents ignored the FDA review (which was their own exhibit) and its manifest inconsistency with Lisse (2003). The review does, perhaps, throw a little light on the “terrible situation” into which, according to Dr Scolnick, the marketers had placed MRL as a result of insisting that the ADVANTAGE trial should be conducted.
658 Dr Reicin was cross‑examined about an email she received in April 2000 in the context of then preliminary results of advantage to which she was blinded, but responded by relying on the final data as reported in Lisse (2003). She relied particularly upon the fact that there were six strokes, all ischaemic, in the naproxen arm and none in the Vioxx arm, adding “if Vioxx had been prothrombotic I certainly wouldn’t have expected to see statistically fewer strokes on Vioxx compared with naproxen.” She was not cross‑examined about the inconsistency between the figures on which she relied and those on which Prof Zipes relied. Neither was she challenged on the following evidence given in her statement in reply:
Finally, ADVANTAGE compared Vioxx to naproxen. Even if we had observed a meaningful difference between Vioxx and naproxen, it would not have materially changed my belief about Vioxx’s cardiovascular safety – we had already seen a statistically significant difference between Vioxx and naproxen in the VIGOR study.
659 In the result with respect to advantage, I am not prepared to hold that it put Merck on any greater notice as to the potential cardiovascular risk of Vioxx than it already had as a result of the VIGOR trial. The comparator in both cases was naproxen, and the highest the applicant can put it is that advantage was consistent with VIGOR. If there were some sound scientific basis upon the strength of which Merck ought reasonably to have viewed the advantage results as taking the matter further, it was put neither to Dr Reicin in cross‑examination nor by the applicant in his final submissions. I could not find, therefore, that there was.
660 The only other series of events in 2001, to which I should refer is the exchange of correspondence involving Dr Scolnick in September and again in November of that year (see paras 172‑174 above). I propose to deal with the timeliness, and with the circumstances generally, of the cardiovascular outcomes study later, but I mention here the applicant’s submission that the correspondence demonstrated that Dr Scolnick was far from satisfied with the safety of Vioxx and most reluctant to accept it as self‑evident that the VIGOR results could be explained by the naproxen effect. It was submitted on behalf of the applicant that –
Dr Scolnick was conspicuous by his absence from the witness box. He was head of [MRL], he was the most senior person involved; he was consistent in his concern to conduct a large study directed to [cardiovascular] outcomes from the moment the VIGOR results were unblinded. It should be assumed that his evidence would not have helped Merck’s case.
The difficulty with this submission is that it omits any identification of the additional evidence that Dr Scolnick would have given had he been called. I accept that, on the evidence as it stands, Dr Scolnick was consistent in his concern to conduct a large study directed to cardiovascular outcomes, although I think that to add “from the moment the VIGOR results were unblinded” somewhat overreaches on the part of the applicant. I accept that it is proper to infer from the correspondence that Dr Scolnick had ongoing concerns in the second half of 2001. It was consistent with the data then available to him that he should have had those concerns. I would not, however, regard the existence of his concerns as bespeaking the existence of facts additional to those given in evidence in the case or as altering the conclusion otherwise proper to be reached on the strength of all the information then available to Merck.
661 In the context of the correspondence of November 2001, it was submitted on behalf of the applicant that Dr Scolnick proposed the conduct of a trial that would test the cardiovascular benefits of naproxen. I have rejected that as an interpretation of his email of 21 November 2001, but on any view he did propose a trial in which Vioxx plus a known or hypothesised cardioprotective agent was set against naproxen alone (plus a pump inhibitor in each arm) in a sample of patients known to have some level of cardiovascular risk. It was put to Dr Reicin in cross‑examination that such a trial could and should have been conducted. She rejected the idea, and pointed out that she had done so in her email of 28 November 2001. In her evidence, she said:
Naproxen versus aspirin, no, that was the point. That was exactly the point. If you are taking patients who need to be on aspirin and you put them into a study evaluating naproxen versus aspirin, those patients who are on aspirin who get put on to naproxen will be taken off their aspirin. So they will instead of getting aspirin which they could forget to take … and still have the antiplatelet effects, they will be given naproxen which they have to remember to take twice a day. They will have greater [gastrointestinal] toxicity because of the naproxen and we didn’t think that that was a study that physicians would be anxious to do or to put their patients in.
The matter was not taken further by counsel for the applicant. Notwithstanding Dr Reicin’s explanation, it was submitted on behalf of the applicant that there was “no evidence that Merck ever determined whether such a study was possible”. If this submission is intended to imply dereliction, I could not accept it. The feasibility of taking patients off aspirin in order to assess the effect of naproxen was rejected on ethical grounds by Dr Reicin, and inferentially her approach held sway at MRL during the period in question. There was, in my view, nothing in that approach that departed from those reasonable standards of prudence and care that the law would expect of Merck in the circumstances then confronting it.
662 Taking all the events of 2001 into consideration, I am of the view that the conclusion expressed in para 634 above with respect to the end of 2000 would need to be amended in some respects only. That relates to the data showing the confirmed CVT mortality from the Alzheimer’s studies (see para 640 above). I refer here to my conclusion that those data ought to have been seen as a conspicuous signal of CVT risk. They gave emphasis to the view which Merck ought to have taken as a result of VIGOR – that there was a worrisome signal of potential cardiovascular risk. They ought to have been regarded as an element in the overall fabric of data then available to Merck which tended to make that signal a realistic one. However, they still did not stand as the whole of that data. In the context of all the information, I do not consider that the position occupied by Merck at the end of 2001 was one in which it ought to have known – ie to have concluded – that the consumption of Vioxx caused or contributed to CVT events generally.
2002
663 Subsequently to about the end of 2001, it appeared that a deal of effort went into the design of the proposed VALOR trial. In his final written submissions, the applicant contended that Merck had designed “but failed to conduct” the VALOR study. Without saying so in terms, the sense of the submission was that Merck was derelict in its abandonment of VALOR. However, Prof Celermajer and Dr Reicin were not cross‑examined on their evidence to which I referred in para 176 above. It was not put to Dr Reicin that she and others at MRL were remiss in not having pursued the VALOR option, or that the reasons for the abandonment of that option articulated by Prof Celermajer were not reasonable ones. In the circumstances, I cannot accept the applicant’s implicit criticism of Merck as to the abandonment of VALOR.
664 There was nothing in the third pooled analysis of May 2002 that had the potential materially to change the position confronting Merck from that which existed in 2001. The new data which were included in that analysis, limited though they were, continued to justify Merck’s contention that no signal of CVT risk appeared. I consider that the thinking reflected in the discussion section of the analysis (see para 183 above) was justified by the data.
665 Weir (2003), which was substantially the work of scientists at MRL, was submitted for publication in the same time‑frame as that in which the third pooled analysis was being prepared. Prof Woodward’s comment on this article included the following:
The enlarged data add support to Reicin et al’s conclusion of no effect but still do not approach the critical size to rule out a chance finding of no effect due to an insufficient sample size.
Save to rely on Prof Woodward’s evidence, and to reiterate his reliance on Prof Madigan’s approach to the arthritis and Alzheimer’s data (which I have rejected), the applicant’s submissions made no point about Weir (2003).
666 2002 was a significant year in Prof Harper’s assessment of the information by reference to which a scientific conclusion that Vioxx was most likely pro‑thrombotic could, and should, have been made. As mentioned in para 594 above, Prof Harper joined in the opinions of Profs Celermajer and Vaughan that the VIGOR results were “hypothesis generating”. By the end of 2002, however, he considered that there were sufficient data for Merck to have realised that Vioxx “had significant cardiovascular toxicity”. This opinion was based substantially upon emerging evidence as to the cardioprotective effect of naproxen. Prior to 2002, Prof Harper held the view that naproxen might, in an extreme case, have had a cardioprotective benefit something akin to that of aspirin – about 30%. However, the myocardial infarction relative risk figure of 5 revealed by the VIGOR data meant that, to stand as a complete explanation for that result, the cardioprotective benefit of naproxen would have had to have been in the vicinity of 70%. Prof Harper continued:
I think there were seven studies, some observational, some case control studies of naproxen, which clearly showed that naproxen was not having that sort of beneficial effect. So that’s why I have said at the very least, by the end of 2002, it should have been known that the initial explanation of the VIGOR study was incorrect and that there was an important signal that Vioxx had … a cardiac toxicity.
Prof Harper here referred to studies which had been published as separate entities in 2002, but which were brought together in Juni (2004). The analysis was similar to that undertaken by Prof Woodward and mentioned at paras 351‑353 above.
667 Prof Harper accepted that the studies mentioned by Juni and colleagues were observational ones, and that none of the published works contained any statement about the levels of compliance achieved in them. Neither they could, of course, since the researchers concerned were not in a position either to influence or (contemporaneously) to monitor compliance. I have referred (at para 366 above) to Prof Woodward’s acceptance of the proposition that, standing alone, Watson (2002) would “shine a light that said naproxen was cardioprotective”. I consider, therefore, that, had Merck looked at all the published naproxen studies as at the end of 2002, it would have noted that they were observational ones, it would have concluded that rates of compliance could well have been variable, it would have been aware that two of them (Watson and Schlienger) contained relative risk intervals that were consistent with there being as much as about a 60% cardiovascular benefit from the consumption of naproxen, and it would not reasonably have been driven from the view which it then took that the cardioprotective effect of naproxen was responsible at least for a major part of the myocardial infarction differential in VIGOR.
668 The position as at the end of 2002 was, in my view, no different from that which confronted Merck a year earlier, and as to which I expressed my conclusion in para 662 above.
2003
669 Prof Carlo Patrono, described by the applicant in his submissions as “an Italian pharmacologist widely regarded as a leading expert in the field of prostacyclin‑thromboxane effects”, was a co‑author of Baigent (2003), published in January 2003, and tendered by the applicant. It contained a section headed “Coxibs and cardiovascular disease”. The authors referred to VIGOR, and identified the cardioprotective effect of naproxen and the promotion of thrombosis by Vioxx as the two mechanistic explanations which had been proposed for the myocardial infarction results of that trial. I have set out (at para 375 above) what they said about the former. As to the latter, they considered first the possibility that the result might be explained by an alteration of the ratio of endothelial prostacyclin to platelet‑derived thromboxane. This was, of course, the FitzGerald hypothesis. Their conclusion on this aspect was:
It seems unlikely, therefore, that alteration of the homeostatic balance of endothelial PGI2 and platelet‑derived TXA2 could explain the higher risk of myocardial infarction among patients allocated to receive rofecoxib in the VIGOR trial.
The authors then considered the possibility of a blood pressure effect, noting that the non-selective NSAIDs and coxibs were “known to induce an increase in blood pressure through their effects on renal prostaglandins”. They concluded that differences in blood pressure could not explain much of the excess of myocardial infarctions observed in the Vioxx arm of VIGOR. Their general conclusion was:
Therefore, although the cause of the apparent excess risk of myocardial infarction in the VIGOR trial cannot be conclusively established, a combination of some cardioprotective effect of naproxen and chance does seem to offer a plausible explanation for these unexpected findings. Although other mechanisms cannot be discounted, there is currently little evidence in humans to support a prothrombotic effect of coxibs.
There is, in this passage, no support for the contention that, at about the start of 2003, the state of scientific understanding was such that Merck ought reasonably to have concluded that Vioxx caused or contributed to CVT disease. Quite the contrary.
670 Protocol 078 was completed in April 2003. My attention was not drawn to evidence of the final CVT or APTC relative risk figure arising from this study as such (ie apart from the overall figures for the three Alzheimer’s disease protocols), but there were 38 CVT events within 1361 patient‑years in the Vioxx arm (a rate of 2.79 events per 100 patient‑years) and 36 such events within 1551 patient‑years in the placebo arm (a rate of 2.32 events per 100 patient‑years). Of these data, the members of the study group for the protocol said in Thal (2005):
Elderly patients are at increased risk for serious vascular events. Rofecoxib did not appear to increase the risk in this study, since the number of confirmed serious thrombotic vascular events was similar in each treatment group.
The authors used the expression “elderly patients” because the median age of the patients in the study was 75 years. The completion of Protocol 078 in 2003 also gave final form to the Alzheimer’s disease mortality data. As mentioned in para 446 above, over the three studies there were 12 CVT deaths in the Vioxx arms and seven in the placebo arms. Relevantly, these results were broadly in line with the 2001 interim data to which I referred in para 642 above. The conclusion I expressed on that occasion remains valid for 2003.
671 It was not until 25 July 2003 that Merck submitted the final version of Protocol 203 to the FDA. That was the cardiovascular outcomes study that began to be the subject of consideration within MRL in late 2001. Although the applicant made a number of criticisms of Merck in relevant respects, tardiness between March 2000 and late 2001 to commence planning for a cardiovascular outcomes study was not one of them. Dr Reicin did not, in chief, explain how that delay (if delay it be) came about, and she was not cross‑examined on the point. Subsequently, there was seemingly some time spent on the design of the proposed VALOR study, which was abandoned in March 2002, and then upon the design of the study which became Protocol 203. The first version of that study was submitted to the FDA in October 2002, and consultations apparently followed as outlined by Dr Reicin in her evidence referred to at para 176 above. Although the time intervals seemingly tolerated by this process of consultation with the FDA may perhaps be viewed as inconsistent with a perception of the pressing importance of testing the hypothesis of cardiovascular safety, Dr Reicin was not challenged on this aspect under cross‑examination and I think that nothing ultimately turns on it.
672 I express the latter view because, assuming always that the design of Protocol 203, even if completed much earlier than it was, would have included APPROVe, VICTOR and ViP, it was not the conclusion of Protocol 203 itself that ultimately caused Merck to withdraw Vioxx from the market. It was the emergence of the statistically significant excess of CVT events in the Vioxx arm of APPROVe. That trial commenced in early 2000, before the VIGOR results were available. If the applicant intended to propose that there was some way of conducting a large placebo‑controlled cardiovascular outcomes study that would have yielded a meaningful result before September 2004, he articulated the details and architecture of such a study neither in his submissions nor in the cross‑examination of Dr Reicin. It must be remembered that placebo‑controlled studies are problematic in populations of patients that require drugs for therapy. Dr Marks said that a placebo‑controlled RCT was preferable both for sponsors and for regulatory agencies, but added that –
… if the study were to run for two years, the use of a placebo group might be unreasonable for ethical reasons because all subjects would require an active drug for their condition, so would require a positive control as the comparator.
Similarly, Prof Celermajer said:
[The] area of trials in arthritis does not lend itself easily to placebo‑controlled trials, as patients with arthritis require some medication for pain relief and the use of placebo, especially for long durations, may therefore be considered unethical.
There is, in the circumstances, no basis in the evidence for me to second‑guess Merck about the design of Protocol 203, the result of which is that, whatever tardiness might have been evident in developing and implementing that design, it was the duration and data of APPROVe as an entity within the study that ultimately provided to Merck the evidence of cardiovascular risk that was decisive.
673 In his written submissions, the applicant sought to give relevance to certain circumstances which existed in May and November 2003. The point related to the APTC relative risk figures as they appeared to the unblinded External Safety Monitoring Board for the APPROVe trial in those months. The applicant pointed to the following passage in the minutes of the Board’s meeting on 17 September 2004:
The trend for excess risk for treatment B for confirmed APTC events has continued to grow at each meeting over the last 1‑2 years. In May 2003, the hazard ratio was 1.2; in November 2003 it was 1.4; last February it [sic] 1.8; and currently it is 2.2. Whereas there was an excess of 2 events on treatment B in May 2003, there are now 17.
While the applicant acknowledged that Merck did not have access to the figure as it existed in May and November 2003, he submitted that, if Merck had taken reasonable action to increase the sensitivity for CVT events in its studies, or advised its monitoring boards to change their protocols, Merck would have had this information. I must say that I have no idea what the applicant meant by either of these suggestions. The subject was not canvassed with Dr Reicin while she was giving evidence. Neither was it put to her that at some unspecified point during the conduct of the APPROVe trial, Merck should have arranged for “interim unblinding” with respect to the emerging data, as it had done, in a limited way, with respect to the emerging Alzheimer’s disease data in the aftermath of the VIGOR results. Accordingly, I have no idea how Dr Reicin would have reacted to such a suggestion, nor what it would have involved in the context of Protocol 203 generally. Absent the assistance of Dr Reicin’s evidence in relevant respects, I do not consider that I am in any position to uphold these essentially technical criticisms of Merck in relation to the APPROVe data as they were known only to the Board before September 2004.
674 I do not believe that Merck’s position as at the end of 2003 was materially different from that which had obtained a year (and two years) earlier. The conclusion I expressed in para 662 remains valid for December 2003
2004
675 In March 2004, Capone (2004) was published in Circulation. The passage from it which I have set out above at para 367 provided some independent scientific support for the view which Merck then took as to the role of naproxen in the VIGOR results. I should not, however, leave the article without setting out the authors’ closing paragraph:
In conclusion, the present study demonstrates the pharmacodynamic plausibility of a COX-1-dependent cardioprotective effect of naproxen and contributes to the interpretation of the VIGOR cardiovascular findings. Although our results are mechanistically informative of what may happen under the best‑case scenario of a randomized clinical trial of regular, prolonged use of a high‑dose reversible COX inhibitor, practicing physicians should not assume that the same holds true in the less‑than‑ideal circumstances of real‑life use of these drugs, which is neither regular nor continuous nor necessarily at high doses.
I read this as suggesting not that naproxen in fact explained the VIGOR results, but rather than it plausibly contributed to an explanation of them.
676 Also, in March 2004, Merck forwarded the fourth pooled analysis to the FDA. Although the relative risk was not significant, and the text accompanying the analysis contained the view that “the data continue to support the lack of a prothrombotic effect with rofecoxib”, the point estimate of the relative risk was now 1.26. It had risen from 1.04 in May 2002. The lower point of the 95% confidence interval had risen from 0.72 to 0.89. This drift, which accompanied the addition of more data in the analysis, ought to have been of some concern to Merck. However, I do not believe I could put it higher than that, and the applicant did not base his case on the pooled analysis of March 2004. As I have said, his case was based (in relevant respects) on different data altogether – those derived by Prof Madigan which I have declined to accept.
677 A final event of interest was the publication in September 2004 of Patrono (2004). I have, at para 385 above, set out a view which the authors of that study expressed as to the explanation for the VIGOR results, on which Prof Celermajer relied. It will be seen that it was very nearly the same sentiment as expressed by Prof Patrono in Baigent (2003), cited at para 669 above. As I did with that article, I am disposed to view Patrono (2004) as implying the absence of any established scientific consensus that Vioxx was pro‑thrombotic.
678 All things considered, the applicant’s case against Merck relies heavily upon two co‑operating propositions: first, that Merck should have regarded the FitzGerald hypothesis, rather than the naproxen hypothesis, as the “correct” mechanistic explanation for the results of VIGOR; and secondly, that Merck should have viewed the data from its own arthritis and Alzheimer’s disease studies as Prof Madigan did rather than as it actually did at the time. For reasons which I have attempted to explain, I am not persuaded that it was unreasonable of Merck not to have acted consistently with these propositions. Once it be accepted that it was reasonable for Merck to have regarded naproxen as a potential contributor of some significance to the VIGOR results, to have applied the CV SOP to the collection and analysis of data from its clinical trials, to have pooled those data as it did, and to have regarded the FitzGerald hypothesis as itself as the subject of some uncertainty, as I do, I could not hold that there was any time before September 2004 when Merck ought reasonably to have taken the view that Vioxx was – as distinct from might be – pro‑thrombotic.
Summary
679 That brings me back to the opinions of the cardiologists, which have been of central importance in this aspect of the litigation. Here I return to para 594 above, which dealt with the position immediately after the release of the VIGOR results. Commencing with Prof Zipes, for reasons already given I do not accept so much of his opinion as was based on the re‑analysis of the arthritis data carried out by Prof Madigan. The analysis by Dr Chen to which Prof Zipes referred was that mentioned in para 637 above: it related to all‑cause mortality. I have dealt with that aspect by accepting Merck’s close investigation of the circumstances surrounding the deaths in question. The Juni study mentioned by Prof Zipes was in fact published in November 2004, after Vioxx had been recalled. And the internal memorandum of Dr Shapiro was, as I have said, wholly consistent with the approach which Merck took to its data generally, and of which I have not been critical. In the result, I am unable to accept Prof Zipes’ opinion that Merck ought, in 2000, to have accepted that Vioxx had significant cardiovascular toxicity.
680 Prof Harper would have regarded the VIGOR trial as no more than hypothesis‑generating until, by about the end of 2002, there was sufficient published research to permit the conclusion that the results of VIGOR could not be explained by the effect of naproxen – see para 666 above. However, for reasons I have given, I take the view that, looking at the matter as at the end of 2002, Merck would reasonably have concluded that naproxen had the potential to be as cardioprotective as aspirin, if taken regularly. There was no time, subsequent to that but before September 2004, when Merck ought to have taken a different view.
681 That leaves Profs Celermajer and Vaughan. They joined in the opinion that the VIGOR results presented a worrisome and important signal of potential cardiovascular risk that mandated comprehensive analysis and investigation. I accept that evidence of the cardiologists. I also accept that Merck did give the problem the analysis and investigation that was reasonably required in the circumstances. For reasons I have given, I consider that Merck was entitled to regard the generality of the placebo‑controlled data available to it as providing little or no confirmation of the VIGOR results. Neither, however, did they remove the cause for concern to which the cardiologists unanimously referred. That was recognised by the scientists at MRL, most conspicuously by Dr Scolnick himself. He was amongst a number of those scientists who keenly felt that underlying sense of concern, notwithstanding those data. Thus the origins of Protocol 203. It was, I consider with respect (and here I speak with the considerable advantage of hindsight), both prudent and wise to include APPROVe in that study. It meant that, to an extent at least, the time which had passed since the VIGOR results became available was not wasted. APPROVe was a major placebo‑controlled RCT which had the obvious potential to be about as informative as any individual study could be, given the constraints applying to such trials in populations of patients experiencing pain.
682 Thus I reject the applicant’s case that, before late September 2004, the respondents knew or ought to have known that Vioxx caused or contributed to the pleaded cardiovascular conditions. I accept his alternative case that the respondents should have regarded the VIGOR results as a worrisome and important signal of potential cardiovascular risk. I reject the applicant’s case that Merck proceeded as though no such signal existed. It recognised the signal and sought, within the constraints to which I have referred, to confirm or deny, as a matter of reality rather than of potentiality, the existence of the risk implied thereby.
part Vi − the applicant’s own circumstances
The applicant
683 The applicant was born on 28 June 1950. He played sport regularly as a boy. He and his brother were keen cyclists, and were members of the Northcote Cycling Club. At the age of 13, he contracted hepatitis A after swimming in the Merri Creek. He suffered from migraines in his youth, and continues to do so today.
684 The applicant left school in 1965 at the age of 15. He applied to join the Navy, but was not then accepted. For the next three years, he had a series of jobs including as a projectionist at the Curzon Theatre in Melbourne, at Maxwell’s Radio in Elizabeth Street, and as a typewriter mechanic.
685 When the applicant turned 18 he again applied to join the Navy, and was accepted on 11 January 1969 as a medic. He worked as a Navy medic for about 3‑4 years, during part of which time he also attended St Lucia’s University in Queensland to study pathology. He worked in medical and surgical wards at HMAS Cerberus and on various other ships as a sick berth attendant. He did a 9‑10 month medics’ course at HMAS Cerberus, and later worked as an instructor at the Navy medical training school. His work mainly involved studying areas such as trauma, recognition of diseases, tropical diseases, hygiene, anatomy and physiology. He was then classified as an Able Seaman – Medic.
686 The applicant was chosen to work in a pathology laboratory, doing haematology. He learned how to perform blood tests and other pathology tests. In the course of performing this work, he gained an awareness of cardiovascular issues and came to understand the signs of heart problems.
687 In the applicant’s last couple of years with the Navy there was a focus on the dietary content of the meals. He had earlier developed a habit of eating healthily (after suffering from hepatitis A), so dietary focus was not a major issue for him, but, together with his training as a medic, it reinforced the importance of good diet and health. The applicant was a regular smoker when he was in the Navy. He usually smoked a pipe (a pouch of tobacco would last for about a week), but, when he used cigarettes, it was at a rate of about 15‑20 per day. He progressively reduced his use of tobacco between 1975 and 1980, when he stopped smoking.
688 The applicant left the Navy on 20 January 1974 (although he continued serving in the Navy Reserve until 20 January 1979). He then worked for the Board of Works in Melbourne as a driver and trades assistant for a crew that performed maintenance on pumping stations in the south‑eastern suburbs. There is no evidence as to when the applicant left the Board of Works, but his next job was as National Scientific Representative for Terumo Australia, a company which manufactured medical disposables. He was involved in the training of hospital staff in the use of equipment. His employer was the first to introduce portable foetal heart monitors in Australia, and he assisted in training hospital staff in their use. After working for Terumo for about three years, he left that job because it required too much travel.
689 The applicant worked for himself until about 1980, researching the commercial fishing of sea urchins. He obtained an experimental permit to harvest sea urchins for the Japanese and Korean markets. He entered into a contract with a Japanese corporation, Mitsubishi, worth about $4 million annually to catch and process sea urchins and export them to Japan. The permit was due for renewal in November 1985, but the applicant was advised that the Victorian government intended to undertake a three‑year study into the effect of sea urchin harvesting, and the permit was not renewed. As a result, the applicant lost his business and had to sell his boat and refinance his house to survive.
690 Then, which was 1985, the applicant did casual work as a chemical formulator for a friend at a company named Soltech. Soltech was later bought by Ensign Laboratories, at which the applicant became Safety Manager and Manager of Special Gases. Most of his work with Ensign involved dangerous and highly explosive gases, including propane, butane and ethylene oxide. He became very aware of safety issues, and was the company’s first Safety Manager. He remained with Ensign until about 1997, developing a good relationship with the Managing Director. He completed an Associate Diploma in Applied Science and Occupational Health and Safety at the Holmesglen College of TAFE, and a Diploma of Applied Science in Environmental Management at Holmesglen and Dandenong TAFE. These were his first formal qualifications.
691 The applicant left Ensign because he was offered a job as a safety officer at the Williamstown Naval Dockyard with Transfield Defence Systems, later called Tenix Defence Systems. After three months he was promoted to the position of Safety Coordinator. His work for Tenix required him to start before 6.00 am, to inspect all the ships in the yard prior to 7.00 am, to conduct compartment inspections of the ANZAC frigates, and to carry out safety audits and accident investigations. While at Tenix, the applicant significantly improved the safety record on the ships and in the dockyard, reducing the accident rate quite considerably. When he started, there were about 18‑20 lost‑time injuries per month; by the time he left, these had been reduced to 2‑3 per month. He had a medical check‑up performed while employed at Tenix. This did not identify any particular problems. He was told to keep an eye on his blood pressure, but he did not believe that his blood pressure was a significant problem. He was not prescribed any medication for it. He also had cholesterol and triglycerides tests performed on his blood. He believed the results were normal. In 1999, the ANZAC frigate construction program on which the applicant had been working was winding down, and he left with a redundancy package.
692 Some months later, the applicant was offered a job with BHP as global safety manager of its marine fleet of 26 ships. To obtain this job, the applicant was required to pass a medical test. The job paid about $75,000 per year. The applicant was based in Melbourne, but the job involved a lot of travel. This meant that he could not keep up with his regular cycling, although he made a point of using the hotel gyms and the exercise equipment on the ships when he was away from home. Every twelve months when he was with BHP, the applicant had a “cardiac workup”, which involved undertaking exercise to get his heart rate up to 180 beats per minute for fifteen minutes, and then timing how long it took for it to return to normal. The applicant remembers a doctor telling him that his cardiac recovery was excellent. In 2001, BHP’s marine fleet was put into a joint venture with Teekay Shipping, and the applicant transferred to the new company. However, Teekay closed its Melbourne office about a year later, and the applicant was offered the choice of transferring to Vancouver or to Sydney. He agreed to work in Sydney, but he felt that Teekay’s commitment to safety was not as good as BHP’s and, after giving approximately 12 months’ notice, he left Teekay on 2 October 2003, intending to set up his own consultancy.
693 On 3 October 2003, the applicant caused a company called “Graeme Peterson & Associates Pty Ltd” (“GPA”) to be incorporated. This was to have been the vehicle through which he would conduct his consultancy business, namely, the provision of occupational health and safety solutions, especially for the maritime industry. An aspect of the business was to have been the undertaking of safety audits, inspections and training.
694 While working in maritime safety at BHP and Teekay, it became apparent to the applicant that there was no way safely to shut down the energy systems in some parts of a ship, such as would be required during maintenance, for example. He developed a system to identify, isolate and lock down a particular system or piece of equipment, which he called “Lockout”. In his evidence, the applicant described this system as follows:
The basis of Lockout was to enable securing of all isolators in any mechanical, electrical, hydraulic or any other energy system, to be able to secure those isolators with a device, whatever was necessary – a clamp or whatever it took – secure that with a lock. The lock is then placed in a keyboard that has a clear front on it. The keys and the locks are all engraved with identifying numbers. There's tags that are attached to the lock and a part of the tag is also left where the lock has been drawn from, so there is a double-check of where the lock is. They are serial numbered to give it a traceability. The person in charge of the job then, once all those devices are locked and secured and tested, he then locks the door of the board, the clear door, so all the keys are visible and he has locked it with another lock. They are colour-coded, all the isolation locks are yellow. His key for securing the board is green. It is also engraved and he is the custodian of that key. Everybody that then comes to work within what we call the isolation barrier, within the area that has been isolated, has their own personal lock which has another engraved identifier and key and they also place them on the door of this board so if you have six people working on the job you have there six red locks, you have the isolation coordinator's green lock and they are all - the whole seven of those locks - securing the keys to the isolation yellow locks inside there. So until everybody has left the job, left the isolated area or the barrier nobody can get to the keys without physically breaking open the board. So once people are finished their jobs, they remove their lock, take it with them. When the isolation coordinator is satisfied that everybody is finished he checks that the personal safety locks have been removed, that the area is clear. He opens the board, takes the keys for the isolators and de-isolates ….
In 2003 the applicant won the “SEACARE” award for the best solution to an occupational health and safety problem in the maritime industry for his “Lockout” system. The system is now fitted to 180 ships worldwide.
The applicant’s back pain
695 While in the Navy, the applicant commenced to experience back pain. That has remained with him ever since then, to a greater or lesser extent. At least by 1983, he had commenced taking anti‑inflammatory drugs for this condition (he thought it may have been earlier than that). In that year, he was prescribed a medication called Indocid. Under cross‑examination, he accepted that he had subsequently taken that and other drugs called Clinoril, Voltaren, Feldene and Orudis SR200, in each case to obtain pain relief with respect to his back. He accepted a series of propositions put to him by counsel for the respondents: that the sort of jobs he was doing in the ’80s and ’90s required him to bend, to climb ladders, to get into confined areas and to walk around ships and other like areas and spaces; that that caused him to spend a lot of time on his feet, it involved a lot of bending and twisting as he went about his duties, and those activities exacerbated his back pain; and that the reason he took the drugs was to enable him to keep working without pain. The applicant said that, in the 20 years before he commenced taking Vioxx, he had been taking NSAIDs regularly.
696 The applicant said that he experienced some very bad gastrointestinal effects from his anti‑inflammatory medication. He suffered from very painful gastric reflux which sometimes caused him to vomit. He would take Mylanta and Maalox, and peppermint, to prevent or to reduce the gastric pain caused by the anti‑inflammatory drugs which he took. The applicant also agreed with counsel for the respondents (who was cross‑examining by reference to medical records) that he had had a number of investigative procedures in this regard. In 1985 he had a barium meal examination. In the same year he had a gastroduodenoscopy, which found a degree of inflammation in his stomach. He continued to suffer from indigestion, and was prescribed a drug called Pepcidine. In 1995 he was prescribed a drug called Losec, which he has taken regularly ever since. That was, and is, principally because of gastrointestinal problems. In 1993 he had a gastroscopy, which demonstrated some mild to moderate inflammation of the oesophagus. He had a further gastroscopy in 1994. In 1997 he had both a gastroscopy and a colonoscopy, which showed some abnormality in his gastric area. He had another colonoscopy in 2002 because of the possibility of bleeding.
697 It was explained to the applicant at the time, and he understood it to be the case, that these gastrointestinal problems were associated with the anti‑inflammatory medication that he was taking. Over the 20 years that he took NSAIDs, that period, the applicant was balancing in his own mind the issues of being able to work without pain and of putting up with such symptoms as he had. He said that, in consulting with his doctor about a drug, he would try to find a balance between a good anti‑inflammatory effect and a low level of gastric discomfort.
698 The applicant referred to some alternative medications that he was taking – fish oil, glucosamine sulphate and “extract”‑type medications, such as shark cartilage and green‑lip mussel extract, for example. He said that he would always read up on these kinds of medications, and would not take them if he was concerned about their contents or side‑effects. He had a practice of taking care to ask about the effects of taking new drugs, and, he said, would always weigh up these effects against joint pain and stomach upsets. The applicant said that he considered himself to be a fairly stubborn person who preferred to reach his own “conclusions about things”, rather than accepting someone else’s word. He considered himself a cautious and safety‑conscious person.
Dr Dickman
699 Dr John Dickman is a general practitioner of considerable experience. He graduated from the University of Melbourne in 1966, and has been in general practice since 1972. He is a member of the Royal Australian College of General Practitioners. He practises at the Tower Hill Medical Centre in Frankston, and has done so at all times which are presently material. He sees about 200‑250 patients a week on average. He first met the applicant in 1970, when they were in the Navy together, and has known him since then. He commenced treating the applicant in 1999.
700 Dr Dickman said that, as a general practitioner with a busy and varied practice, in an average week he would prescribe over a hundred different drugs. Consequently, he had limited time to research the effect of each drug. There were, however, some drugs that he prescribed more regularly than others – they made up his “formulary”, as put to him by counsel for the respondents. He said that, between 2001 and 2004, Vioxx was one of those. He prescribed it “fairly frequently”.
701 From many years of practice, Dr Dickman was aware that a prescription medicine differed from an over‑the‑counter medicine in that it required a medical practitioner’s authority for a patient to obtain it. This was because of the nature of the medication. He knew that every prescription medication had effects ranging from good ones through to serious adverse ones. He accepted what was put to him by counsel for the respondents that he proceeded on the basis that no prescription medicine was absolutely safe for every patient. In the case of every patient, he had to consider the good and bad effects and make a judgment about whether the medicine should be prescribed for that patient.
702 Often, Dr Dickman became aware of a drug’s side-effects by witnessing them in his patients or by hearing about them in the media. If a side-effect actually occurred in a particular patient, he may have moved the patient on to a different drug, or changed the regime or dosage of the existing drug. If the risk of a side‑effect arose, Dr Dickman would weigh up the risk against the benefit provided by the drug to determine whether it was appropriate to continue prescribing it. If he were to continue to prescribe the drug, he would need to be able to advise the patient of the seriousness of the risk posed by the side‑effect, and of the likelihood of the risk coming to pass. He accepted that this was a process through which he went each time he prescribed the drug. On each occasion, he accepted that, in effect, he went through a process that involved asking himself whether anything had changed since he first prescribed the drug. In some situations, he would carry out a more complete risk benefit analysis, such as when the patient’s condition had changed, or when the drug profile had changed.
703 Dr Dickman accepted that, as time went by, he would build up a picture, across his patients, of the tolerability and effectiveness of a particular drug, and as to its tendency to cause side-effects. That would have applied as much to Vioxx as to any other prescription medication. He accepted that, with respect to prescription medicines generally, he was collecting information all the time, and constantly factoring that information into his decision‑making process. Subject to those qualifications, once Dr Dickman had commenced a patient on a long‑term regimen of a particular drug, he tended not to switch the patient to a different drug unless there were a specific reason to do so. Because he prescribed Vioxx frequently, Dr Dickman was accumulating information on it regularly, and scanning, so far as he could, information that he received to see if there were any reason why he should change his prescribing habits.
704 The sources of information about new and existing drugs upon which Dr Dickman relied included specialists, sales representatives, journal articles and the medical press, other general practitioners, product information and patient feedback. Specialists were a particularly powerful source of information about new drugs. He regarded them as more knowledgeable in their areas of specialisation, and better informed about new drugs, than he could be as a general practitioner.
705 Dr Dickman said that it was usual, when a drug was being launched, for the manufacturer to host a number of educational meetings at which the uses and benefits of the medication were discussed. These usually took the form of an expert in the field talking about the drug or the class to which it belonged. Often, the meeting was over dinner at a local restaurant. Dr Dickman regarded dinners sponsored by pharmaceutical companies as attractive because they tended to be informative, they involved dinner and they offered CME points, which were required to maintain a doctor’s licence to practise medicine. He recognised that specialists who spoke at sponsored dinners were compensated by the pharmaceutical company concerned, and he assumed that they provided unbiased and truthful information about the drug. Such dinners were also the occasion for interaction with his colleagues about the speaker’s subject, and for a free-flowing professional discussion generally.
706 Specialists influenced Dr Dickman’s prescribing habits also through his patients. When a patient returned from a specialist with a prescription for a certain drug, Dr Dickman would take note of the prescription, and generally continue to prescribe that drug to that patient. He would often prescribe the drug to other patients in similar situations.
707 Dr Dickman said that information provided by sales representatives about drugs was “sometimes useful”. He saw sales representatives once a week, on a Thursday morning. He recognised that a sales representative’s job was to increase prescriptions of the drugs they represented, but he assumed that the information given by them was truthful. In his witness statement, Dr Dickman said that, when representatives told him that a drug was safe, he assumed that they were telling the truth. However, under cross‑examination, he accepted that, if a representative said to him, “Here is a prescription medicine – I can guarantee you that it’s absolutely safe”, he would be likely to have regarded it as an overstatement. He accepted that he would not take the representative’s word for it, but would think to himself, “I had better go and have a look at what I can find out about this drug to see what the safety of it is”. He would know that a comment by a sales representative about the safety of a drug had to be placed in the context of all of the information that was available about the drug.
708 Sometimes a sales representative would tell Dr Dickman that a drug was now approved for a different indication, such as when Vioxx was approved for the indication of rheumatoid arthritis. In such a case, when he next came to prescribe the drug, particularly (in this example) if it were for someone with rheumatoid arthritis, he would go back to the Product Information to see what the relevant information was with respect to that indication.
709 When a new drug was marketed, there was usually information about it in the medical press. For Dr Dickman, the most prominent publications were the Medical Observer, Australian Doctor and Family Physician. He read these journals when he was able, consistent with the time demands of his practice.
710 Dr Dickman talked to other general practitioners about their prescribing habits and the results they achieved with drugs. At Tower Hill Medical Centre, the general practitioners tried to hold practice meetings about every month or so. One of the subjects discussed at these meetings was prescribing habits and results with different pharmaceuticals.
711 Dr Dickman used a computerised version of the Monthly Index of Medical Specialties (“MIMS”) to look up a drug’s Product Information. In his witness statement, he said that, as the Product Information could be very lengthy, his practice was not to “read it from start to finish”. Instead, he would call up the sections that were of particular relevance, for example on drug interactions. He also agreed with counsel for the respondents that there were some aspects of the Product Information that were more important than others. He accepted, by way of example, that the indication for a drug was important, as were the contraindications, the appropriate dosage and administration, the precautions and the adverse reactions. He agreed that they were important because they related to why he might prescribe the drug, and what might happen if he did. He accepted that he needed to weigh them up as part of his information about the drug. Thus, although he did not read the Product Information from start to finish, he accepted that he looked at the sections that were of particular relevance.
712 Dr Dickman said that he would not be aware of a change to a drug’s Product Information unless notified about the change. However, under cross‑examination, Dr Dickman accepted that, in his practice, he used a computer program called “Medical Director”, which contained a prescribing module, part of which was information about the drug being prescribed. That enabled him, with the click of a mouse button, to access information instantly about a particular product by reference to headings such as “Indication”, “Contraindications”, “Precautions”, etc. He could do this without having to read the whole of the Product Information. He agreed that this was one of the more easily available sources of information. Indeed, Dr Dickman said that he more commonly used the Medical Director software to have access to the electronic version of the Product Information. He agreed that Medical Director was electronically updated four times a year, but said that it did not give him a note of what drugs had changed; nor of an indication that had changed. The paper version of MIMS, which Dr Dickman still received, was updated three or four times a year. The updating version would contain only those drugs that needed to be updated.
713 Dr Dickman obtained information about Vioxx from the various sources mentioned above, and used this information to assess Vioxx’s appropriateness for the applicant and his other patients. There was a great deal of attention paid to the launch of Vioxx (as there had been with Celebrex) because of the perception that the drug would provide good pain relief for arthritis without the risk of the gastrointestinal effects associated with the traditional NSAIDs. As a general practitioner, that is what attracted Dr Dickman’s interest. The gastrointestinal complications of long‑term NSAID consumption could be quite serious. Dr Dickman mentioned one patient – an older lady – who was taking an NSAID and who had a haemorrhage, as a result of which her blood pressure dropped and she suffered a stroke.
714 In late 1999 or early 2000, Dr Dickman attended a dinner meeting about Vioxx hosted by MSDA. A local rheumatologist – of whom Dr Dickman had a good opinion – spoke of the COX-2 specific inhibitors and of Vioxx in particular, which he endorsed. Dr Dickman learned that this new class of drug had the same or a similar overall profile to NSAIDs in terms of effectiveness and safety, but had also the particular gastrointestinal safety benefit. The rheumatologist said that Vioxx was safer than traditional NSAIDs. His opinion about Vioxx was probably the most significant factor in Dr Dickman’s decision to prescribe it to his patients.
715 Dr Dickman’s clear impression at the time was that the new class of COX-2 specific inhibitors, including Vioxx and Celebrex, had a similar profile of effects to that of the NSAIDs, but had the identified gastrointestinal benefits. He considered that it would have been almost negligent not to have prescribed them. In discussions between Dr Dickman and his colleagues, there was a very real fear that they would have been exposing themselves to negligence claims if they prescribed “an older type of NSAID” to a patient who later experienced a gastrointestinal bleed. Save for the treatment of acute gout, Dr Dickman could not think of any situation in which he would have prescribed a traditional NSAID once the coxibs were on the market.
716 Although Dr Dickman did not recall the specifics of the meetings which he had with Merck sales representatives about Vioxx, he did meet them frequently. He said that there was “intense competition” between sales representatives for Vioxx and Celebrex. He recognised their respective activities as amounting (in the words of counsel for the respondents) to a “sales pitch”. The only real difference between the two which he was able to recall being raised by the representatives was that Celebrex was “a sulphide”, whereas Vioxx was not. Dr Dickman later explained that this was a reference to the sulphonamide chain in Celebrex. It seems that some patients were allergic to sulphonamides, and in such cases he would have chosen Vioxx. Otherwise, he prescribed Vioxx and Celebrex about “50/50”.
717 Dr Dickman did not recall any MSDA representatives mentioning cardiovascular risks in relation to Vioxx. Under cross‑examination, he accepted that this was because he had no recollection of any specific interaction at any meeting with a representative. He also accepted that he gave that evidence without the benefit of knowing of the conversation he possibly had with a representative on 13 September 2001, as set out in the list below. He said that, if a sales representative had mentioned the possibility of cardiovascular risks from Vioxx, he would have discussed the issue with other doctors and specialists, and would have raised the matter with the applicant. Indeed, Dr Dickman said that meetings with MSDA representatives left him confident about the safety of Vioxx. In his witness statement, he said:
During meetings with Merck sales representatives about VIOXX, the sales representatives’ emphasis was primarily on VIOXX’s safety.
Under cross‑examination, he was not challenged on this statement, but acceded to the proposition that a regular theme with the representatives was that Vioxx had a real benefit for his patients because it had a better gastrointestinal safety profile than traditional NSAIDs. The other theme was that Vioxx needed to be taken only once a day, which meant that the patient would not get “breakthrough pain”.
718 In evidence was a schedule of the interactions between MSDA sales representatives and practitioners at the Tower Hill medical centre over the period to which this proceeding relates. It reveals that representatives saw Dr Dickman as follows:
· On 9 January 2001 − Christopher Brown
· On 30 January 2001 − Christopher Brown, noting that Dr Dickman was “very happy with Vioxx”
· On 15 February 2001 − Christopher Brown, noting that Vioxx was “going great guns”
· On 8 March 2001 −Christopher Brown and Kieran McAuley, noting that Dr Dickman used Vioxx
· On 20 April 2001 − Christopher Brown, noting “good push for Vioxx as number 1 choice before other Cox 2 …”
· On 2 May 2001 − Christopher Brown
· On 11 May 2001 − Christopher Brown
· On 14 May 2001 − Marcus Daddo
· On 15 May 2001 − Marcus Daddo
· On 16 May 2001 − Marcus Daddo, noting that Dr Dickman “has used Vioxx but usually @ 25mg” and (as I interpret the document) that Mr Daddo “went through the types of patients best suited to Vioxx”
· On 5 July 2001 − Marcus Daddo, noting “quick otc about the benefits of Vioxx … aware of and using”
· On 8 August 2001 − Christopher Brown, noting “benefits agreed now Vioxx is running 50/50 no real cardio issues yet”
· On 12 September 2001 − Christopher Brown
· On 13 September 2001 − Marcus Daddo, noting that Dr Dickman had “some problems patients querying use of Vioxx with HF and MI’s etc, clarified concerns and moved on to Vioxx promo”
· On 14 November 2001 − Christopher Brown, noting that Vioxx was “getting good grounding but with hearty debate”
· On 21 December 2001 − Christopher Brown
· On 10 January 2002 − Kym McCarthy, noting as follows:
Vioxx key messages, CV competitor issues addressed. All prefer decreased side effect profile of Cox 2’s … ID reasons to choose Vioxx 1st line based on SSS.
· On 27 March 2002 − Christopher Brown, noting that Vioxx was going “ok but still high Nsaids”
· On 23 April 2002 − Kym McCarthy, noting “Vioxx efficacy”
· On 14 May 2002 − Kym McCarthy, noting “Vioxx OTC efficacy”
· On 13 June 2002 −Kym McCarthy
· On 23 August 2002 − Kym McCarthy
· On 12 September 2002 −Kym McCarthy
· On 29 October 2002 − Kym McCarthy, noting “Vioxx med info follow up …”
· On 5 December 2002 − Kym McCarthy, noting “Vioxx key messages”
· On 16 January 2003 − Kym McCarthy
· On 20 March 2003 −Kym McCarthy
· On 25 June 2003 − Kym McCarthy
· On 27 June 2003 − Kym McCarthy
· On 22 July 2003 − Kym McCarthy
· On 18 August 2003 − Kym McCarthy, noting “Vioxx … full detail − reminder benefits over others in class”.
719 Save for Mr McAuley, none of the representatives mentioned above was called. I believe I am able to infer that none of them would have been in a position to deny Dr Dickman’s evidence that their emphasis, in their meetings with him, was on the safety of Vioxx. As for Mr McAuley, he proffered no such denial and provided no elaboration on the meeting which, apparently, he had with Dr Dickman on 8 March 2001.
720 Dr Dickman said that, at the time that he prescribed Vioxx to the applicant, he was not aware that use of the drug was associated with increased cardiovascular risk. He first became aware of a possible increased cardiovascular risk associated with Vioxx at an educational meeting, about a couple of months before it was withdrawn, addressed by a local cardiologist. Before then, he had felt confident in the safety of Vioxx, but the implication from the cardiologist was that there was a greater cardiac risk than he had imagined previously. In his witness statement, he said that, had he been aware of an increased cardiovascular risk from Vioxx, this would have affected his risk/benefit analysis in prescribing the drug. He would have urged caution in the use of Vioxx and, in particular, would have recommended use for brief periods only. When it was put to him that he gave this evidence with the benefit of hindsight, he responded: “I certainly think that my current knowledge can’t be eradicated” from the evidence. He said that he would have warned his patients about any known risks to enable them to make informed decisions about their medication, and would have respected their decisions. He said that the increased cardiovascular risk that he now knows to be connected with Vioxx would certainly have warranted a warning to patients who had been taking the drug.
721 Dr Dickman was shown a copy of the October 2001 newsletter of the National Prescribing Service, which he accepted was an independent body largely if not entirely funded by government, the purpose of which was to provide information to doctors about prescribing habits, drugs and the like. Dr Dickman received this publication. His attention was drawn to the following extract:
Do COX-2 selective NSAIDs carry a risk of thrombosis?
Evidence is accumulating that selective inhibition of the COX-2 enzyme may be associated with prothrombotic effects.
The VIGOR study showed a small but statistically significant difference in the incidence of myocardial infarction between the groups treated with naproxen and rofecoxib (0.1% vs 0.4%)…. This trend has also been observed in a review of rofecoxib studies which includes as yet unpublished data. [Reference given to an FDA publication available online.]
…
It is uncertain whether the observed increase in cardiac adverse events with rofecoxib is valid, whether it is specific to rofecoxib only or COX-2 selective NSAIDs in general, or whether some NSAIDs have an aspirin‑like cardioprotective effect and actually reduce the risk of thrombosis. The practical implications of these observations are:
– those who require low‑dose aspirin for cardiovascular prophylaxis should receive it [Reference given to Cleland (2001)]
– consider low‑dose aspirin as an additional risk factor when assessing the overall risk of serious gastrointestinal toxicity with NSAID use in an individual
– low‑dose aspirin may offset any potential gastrointestinal protective effect of COX-2 selective NSAIDs [Reference given to Lichtenstein (2000)]
– the possible increased risk of myocardial infarction, together with known side‑effects such as oedema and hypertension, indicates that COX-2 selective NSAIDs should be used cautiously in persons with cardiac disease (as with conventional NSAIDs).
Asked whether the statement that evidence was accumulating that selective inhibition of the COX-2 enzyme might be associated with prothrombotic effects reflected his state of knowledge at the time, Dr Dickman said that his feelings were “coloured by what I know now but I think I was probably in a state of naïveté and still believing that this class of drug had lesser risk”. He said that he had no recollection of reading this newsletter, but accepted that it was the type of publication that he would have read. As to the statement contained in the last bullet point set out above, Dr Dickman agreed that, if he had in fact read the newsletter, it was something that would have stuck out as an important piece of information. But he added: “… but again, it’s coloured with today’s [information]”. In this and similar respects, Dr Dickman acknowledged that it was difficult, knowing what he now does, to recall his state of mind before September 2004 about the cardiovascular risks of Vioxx.
722 Dr Dickman was also shown a copy of the June 2003 newsletter of the National Prescribing Service. He accepted that the evidence which he gave about the October 2001 newsletter – as to his receipt of the publication and his tendency to read it – was likewise applicable to this one distributed in June 2003. His attention was drawn to the following:
Thrombotic concerns with COX-2s remain unresolved
We are no closer to adequately explaining why more myocardial infarctions occurred in the rofecoxib‑treated subjects during the VIGOR trial than those taking naproxen. An epidemiological study has suggested that rofecoxib doses greater than 25 mg (such as those used in VIGOR) are more likely to be associated with serious coronary heart disease [Reference given to Ray (2002) “COX-2 selective non-steroidal…”] although this conclusion needs to be confirmed.
Until an appropriate study is performed focusing on cardiovascular outcomes in patients taking COX-2 selective NSAIDs, caution should be exercised when prescribing these drugs in patients at risk of cardiovascular events.
What is accepted is that patients who require low‑dose aspirin for cardiovascular prophylaxis should receive it as the benefits are established. [References given to Cleland (2001) and to FitzGerald (2001)] If such patients require an anti‑inflammatory agent, prescribing COX-2 selective NSAIDs preferentially over conventional NSAIDs is not justified on current evidence. [Reference given] Using more than one NSAID concurrently increases the risk of gastrointestinal adverse effects.
Again, with the benefit of hindsight, Dr Dickman acknowledged that it was difficult to say whether this passage represented his state of mind in 2003. He said that he was “still feeling quite comfortable with Vioxx and Celebrex in the patients [for whom he] was using it”. He accepted that his position at the time was that, to the extent that there were side-effects with these drugs, they were not causing him to refrain from prescribing them.
723 A passage from the Australian Adverse Drug Reactions Bulletin, published in October 2003 by the Australian Adverse Drug Reaction Advisory Committee, was put to Dr Dickman, but he was unfamiliar with it. The passage dealt with the then learning as to the link between the consumption of coxibs and cardiovascular disease, and at one point contained a statement to the effect that the evidence for an association between rofecoxib and a risk of cardiovascular events was “inconclusive and indirect”. Dr Dickman said that, if he were aware at the time of information broadly to that effect, it was not sufficient to influence his prescribing, adding, “we are all coloured by our own experiences and I didn’t have any unpleasant experiences with my prescribing of Vioxx or Celebrex.”
724 Dr Dickman was shown the fourth version of the Vioxx Product Information, approved by the TGA on 14 December 2000 (although identified by counsel as the original version approved on 23 December 1999). He agreed that the “Precautions” section was the sort of topic with which he would have made himself familiar; and that the “Adverse Reactions” section was also something with which he would have had some familiarity at the time he commenced to prescribe Vioxx. It was established that Dr Dickman understood the structure and meaning of the tables and other representations of data in this section. When he first started prescribing Vioxx, he was aware of the range of adverse reactions there set out.
725 In 2001, Dr Dickman had occasion, clinical condition requiring it, to advise patients to take aspirin in a low dose as a prophylactic against cardiovascular risk. Aspirin was understood by him to have some antithrombotic tendencies. His attention was drawn to the statement in the “Precautions” section of the version of the Product Information that was approved on 16 November 2001 that Vioxx was not a substitute for aspirin for cardiovascular prophylaxis because of its lack of effect on platelets. He agreed that, when he was prescribing Vioxx between 2001 and 2004, he understood that it was not a substitute for aspirin because it lacked any effect on platelets.
726 Dr Dickman’s attention was drawn to the paragraph headed “Cardiovascular Effects” in the same section of the November 2001 Product Information (see para 258 above). Although he could not put a specific time on it, at some time between December 2001 and September 2004 he knew the ideas encapsulated by this paragraph. That knowledge was not, however, sufficient to make him “unconfident” in the use of the drug. When asked by the court to reconcile that evidence with his evidence in chief that, at the time that he prescribed Vioxx to the applicant, he was not aware that use of the drug was associated with increased cardiovascular risk, Dr Dickman said that he did not regard the risk mentioned in the “Precautions” section of the Product Information – 1.67 events per 100 patient years – as a particularly large risk in the applicant’s case because he regarded him as a “mild to moderate” cardiac risk. Asked in re‑examination to explain what the “Precautions” section conveyed to him, he answered:
The rate of serious cardiovascular thromboembolic adverse events were significantly lower in patients receiving [naproxen] than in the rofecoxib group. So that it indicates [naproxen] to be a safer drug in terms of cardiovascular events but the overall rate, I would think, with Vioxx is still relatively low.
Asked by the court whether, had he had the relevant text on his computer screen at the time he was prescribing Vioxx, and was conspicuously conscious of it, he most likely would still have prescribed it, Dr Dickman said that he would have discussed it with the patient concerned, explained the risk, balanced it against the patient’s cardiovascular risk, and left the final decision to him or her.
The applicant’s experience with Vioxx
727 The applicant was first prescribed Vioxx by Dr Dickman on 10 May 2001. Dr Dickman had no recollection of any discussion between himself and the applicant at that time about the prescription of Vioxx. From a perusal of the records of his practice, however, he was able to say that the applicant had been complaining of increasing pain in his neck. Some radiological investigations were undertaken, and these confirmed osteoarthritis in the neck. Dr Dickman accepted that the applicant had a long history of back pain, which he (Dr Dickman) associated with osteoarthritis. In May 2001, the applicant was suffering from osteoarthritis from the neck down to the low back. He accepted that, although the applicant’s acute presenting problem was pain in the neck, the prescription of Vioxx would have addressed all osteoarthritic pain in his back and spine. Dr Dickman also accepted that, in the circumstances then existing, had he not prescribed Vioxx or one of the other coxibs, he would have prescribed a non‑selective NSAID. The pain being experienced by the applicant had to be treated by something. Dr Dickman chose Vioxx because “it was attractive from the point of view of [the applicant’s] gastro‑oesophageal reflux”. The dose prescribed was 25 mg daily.
728 According to the applicant, Dr Dickman told him that Vioxx gave good pain relief without causing stomach upsets. Dr Dickman recommended Vioxx, the applicant did not request it. Having known Dr Dickman since his time in the Navy, the applicant regarded him as the kind of doctor who asked questions about new products. He considered Dr Dickman’s judgment to be very sound. The applicant said that he always asked his doctors about the side‑effects of any medications they prescribed. The side-effects that Dr Dickman and he discussed in relation to Vioxx were the gastric side-effects of the NSAIDs, which Dr Dickman did not expect to see with Vioxx. Dr Dickman explained that Vioxx worked differently to other anti‑inflammatory drugs. The applicant recalled that Dr Dickman looked through the MIMS entry for Vioxx on his screen, and he indicated that he and others had looked at and discussed Vioxx at length, and “they did not consider it had any major downsides.”
729 The applicant said that he tended “to try to look into” the medications which he was supposed to take, as he was a safety‑conscious person who did not believe in taking actions without knowing the risks involved. He said that he read “the Vioxx label or product information” when he was first put on Vioxx. By “product information”, he was referring not to the “Product Information” as such, but to the folded sheet of information that was included in the pack in which the Vioxx tablets were supplied. In the lexicon of the TGA, that is the “Consumer Medicine Information”. The applicant did not recall reading anything that suggested or implied that Vioxx would increase his risk of suffering a heart attack. There was nothing which indicated a concern about cardiovascular safety. He said that, had he read anything suggesting that there would be a serious side‑effect like that, he would have “headed straight back to Dr Dickman to ask more about it”. In his witness statement, the applicant said that he did not read the Vioxx product information (ie the Consumer Medicine Information) or MIMS entry each time he received a new Vioxx prescription, but his discussions with Dr Dickman at such times would always include questions (from Dr Dickman) about how he was tolerating the drugs he was on, and questions from him (the applicant) about whether Dr Dickman thought that he should continue using those drugs, and whether anything had changed since the last time they were discussed between them. He said that he “probably would have gone back to re‑read the Vioxx MIMS entry once or twice during this period”, but he mostly relied on his conversations with Dr Dickman and his initial reading of the Consumer Medicine Information to satisfy himself that the pills “were OK to consume”. The applicant was never alerted to any concern about cardiovascular risks involved in Vioxx use until the drug was recalled.
730 While using Vioxx, the applicant did not experience any gastrointestinal effects. He was “feeling quite good for most of the time” he was on Vioxx. He was “even able to go for 20 km walks with our dogs on Sunday mornings”. After he had been using Vioxx for a while, he told Dr Dickman that it was “a great drug”.
731 The applicant was cross‑examined extensively as to his knowledge about Vioxx, and as to his readiness to take Vioxx, notwithstanding being aware of a range of side‑effects, in May 2001 when he was first prescribed the drug. He was shown two documents, neither of which he identified as something he had read at that time. The only familiar aspect of one of them was that it contained a structural representation of the rofecoxib molecule, which the applicant thought he may have seen.
732 The first document shown to the applicant was one which was not tendered, in which circumstances I shall refer only to the applicant’s oral evidence about it. Asked about a “mild” side-effect referred to in the document as “high blood pressure”, the applicant said that, had he read the document, he would not have associated that with a heart attack, since he would not have taken a “mild” side-effect as being indicative of a heart attack. Had he read the document, he would also have noticed that another side-effect mentioned was “pain or tightness in the chest”, which he understood to be a sign associated with the occurrence of a heart attack. He accepted that that would have been a serious side-effect of which he would have been aware had he read the document, but he added that he would also have read a notation to the effect that this side-effect was amongst those “reported rarely in people taking Vioxx and not knowing whether they are definitely caused by Vioxx. Don’t be alarmed by this list of possible side-effects. You may not experience any of them.” However, he accepted that, if that was the document that he read in May 2001, the information in it did not deter him from taking Vioxx. As I have said, it was not established what this document was, nor whether the applicant ever saw it.
733 The other document was the Vioxx Product Information approved by the TGA on 16 May 2001. The applicant was cross‑examined on the basis that this was current at the time that he was first prescribed Vioxx. That was not strictly accurate, since that prescription was written on 10 May 2001, but probably nothing turns on any differences. In the section headed “Adverse Reactions” (which the applicant understood to be the same as “side‑effects”), there was a table listing all adverse experiences, regardless of causality, befalling people who had taken Vioxx in clinical trials of patients with osteoarthritis in which the comparators had been placebo, ibuprofen or diclofenac. Only events occurring in at least 2% of patients receiving Vioxx were included. The applicant’s attention was drawn to the entry for hypertension, in which the figures were 3.5% for Vioxx and 1.3% for placebo. He did not recall seeing these figures at the time. He accepted that, had he seen it, it was not sufficient to give him concern about taking Vioxx, but he added: “… given that hypertension isn’t a heart attack”.
734 The applicant’s attention was then drawn to the adverse events which had occurred in more than 0.1%, but fewer than 2.0%, of patients taking Vioxx (see para 225 above). Under cross‑examination, the applicant was induced to accept that he had a general understanding of the meaning of most of these terms, but only one of which (venous insufficiency) he accepted as being associated with having heart attacks (or, he added, migraines).
735 The applicant was then taken to a passage in the Product Information that dealt with serious adverse reactions that occurred in fewer that 0.1% of patients taking Vioxx (see para 225 above). It was established that the applicant understood these terms. It was put to him that, had he read the document, it would have been immediately apparent to him that there were serious adverse reactions with Vioxx which included strokes, heart attacks and other like conditions. He responded that he would have taken into account the statement that these reactions occurred at a patient‑rate of less than 0.1% in patients taking Vioxx. He said that, had he read this passage, he would have discussed it with his doctor. It was put to him that, given that these events occurred in less than 0.1% of the population studied in the studies concerned, it would not have caused him to refrain from taking Vioxx. His response was that he would have spoken about it with his doctor.
736 The applicant was not shown the new cardiovascular paragraph in the “Precautions” section of the Product Information, as introduced in the November 2001, nor asked how he would have reacted to that paragraph.
737 In his witness statement, the applicant said that, if Dr Dickman had told him of the cardiovascular risk involved with the consumption of Vioxx, he would have “weighed up the positives and negatives about the drug before deciding to take it”. He would have been “pretty wary of it, as joint pain and gastric upsets would not kill me, but a heart attack could”. He said that he would have wanted to know the exact risks involved before making a decision. At the very least, had he known of any risk, he would have discussed it much more with Dr Dickman. If Dr Dickman had given him the impression that there was a cardiovascular risk, he would not have taken Vioxx, even if told that it would also get rid of his gastric pain. The applicant said that, had he understood, before taking Vioxx, what he now understands, he would not have taken Vioxx; that, had he been told that a clinical trial of Vioxx against naproxen had shown that there was a substantially greater risk of adverse cardiovascular events with Vioxx than there was with naproxen, he would not have taken Vioxx; and that, had he been told that taking Vioxx would approximately double his risk of having a heart attack, he would not have taken Vioxx.
738 I allowed the evidence referred to in the previous paragraph to stand only once the applicant had been examined in chief on the subject, as I was concerned about the imprecision, and tendentiousness, of statements based upon the assumed existence of a certain “cardiovascular risk” without the court knowing the extent of the risk, or the increase in risk, which the applicant had in mind as being enough, hypothetically, to have caused him to act differently. In that examination, the applicant said that he would not have taken Vioxx if Dr Dickman had said to him that his risk of a heart attack, whatever it happened to be, would double, or would increase by 40% or 50%. Asked to state the percentage increase in risk of heart attack or equivalent cardiovascular problems which he would have regarded as acceptable, and consistent with which he would still have taken Vioxx with the gastric benefits that it offered, he responded: “I would have had to discuss that with my doctor at the time. As a balance it would have to be a very low risk and it would have to have the other benefits that I was taking the medication for but I certainly would have relied on his advice.” If it came to a choice between naproxen and Vioxx, and the applicant were told that “the risk of a heart attack was two or three or four times as high on Vioxx as on naproxen”, the applicant said:
I would have wanted to know where the figures came from because I wasn’t comparing it with anything else other than if my doctor had told me it was a greater risk than not taking it purely as not taking it. Not taking something else but just not taking Vioxx as against taking Vioxx. I can’t speculate what I would have said about anything else.
The applicant said that his present understanding was that the consumption of Vioxx at least doubled his risk of having a heart attack, “and possibly [made] it 500 per cent more likely”.
739 Under cross‑examination, the applicant accepted that his evidence that he would not have taken Vioxx if informed that it would have doubled his risk of having a heart attack was, or at least may have been, affected by “hindsight bias”, a concept with which the applicant, from his area of work, was well familiar. Indeed, because of the applicant’s training and experience, he was able to respond intelligently to questions put to him in cross‑examination which, to another witness, might have been too technical. It was put to him, in effect, that there was a world of difference between a doubling of risk from 1/10 to 2/10 on the one hand and from 1/100,000 to 2/100,000 on the other hand. He denied that he would, if told that taking Vioxx would double his risk of a heart attack, only have asked himself what the absolute risk was. He accepted that he knew, over many years of taking NSAIDs for his back pain, that such drugs carried with them some risk of quite serious gastrointestinal problems, even to the extent of requiring hospitalization and, in very serious cases, death. Notwithstanding that knowledge, he said that he had never asked his doctor what was the actual the rate of serious gastrointestinal effects involved in the taking of an NSAID. Cross‑examination then proceeded as follows:
In other words, your consumption of that range of medication was without any knowledge of what the actual rate was?---General rate, no, but as far as my risk, I was satisfied that my doctor had considered that.
So you relied on your doctor to consider whether the side-effects of the various NSAIDs that you took were appropriately balanced in your case?---Yes.
Against the benefits?---Yes.
If your doctor thought it was an appropriate balance and prescribed it for you, you took it?---Generally.
740 The applicant was next taken to so much of his evidence in chief as involved evidence that he would not have taken Vioxx if told that it would increase his risk of having a heart attack by 40% or 50%. It was put to him that, for him to have immediately acted on a suggestion that his risk, whatever it was and without knowing what it was, would be increased to that extent would have been contrary to the way in which he practised his business of safety and risk management. While the applicant accepted that, in his line of work, he did usually want to know the absolute level of a particular risk, he rejected the suggestion that, in 2001, he would have taken such an approach to deciding whether or not to take Vioxx.
741 The applicant was asked about his answer in chief that, for him to have been persuaded to take Vioxx against the knowledge that there was some risk, the estimate of the risk would have had to be a low risk, and he would have discussed it with his doctor. He accepted that the decision whether to take Vioxx involved more than a straight comparison of two situations, one in which there was a risk and the other in which there was not. Rather, it was a more complex picture, one in which “a weighing process” was involved, because the hypothetical situation related to a drug which might have increased his risk of heart attack, but reduced the risk of serious gastrointestinal effects. The applicant said that, in such a situation, he would have sought his doctor’s guidance. The cross‑examination continued:
And you feel quite unable now to say what your reaction would have been to the information you got in the course of such a complex process in May 2001. Isn’t that right?---To a certain extent, taking into account, as you say, the bias of hindsight.
It’s not a question you would be prepared to answer now, is it?---I would still attempt to answer it but I would have to weigh what I know now as against what I knew then.
You would accept that if one was attempting to get a snapshot of your mind in May 2001 that snapshot would not be represented by what you could say today?---No, I would say that I would have to try and remember what I was aware of, what I knew, what my impressions of the risk of heart attack and things were then.
That’s not an exercise you have ever done?---Yes, I have.
You have done that?---Yes.
Was that the exercise you were doing when you answered these questions with Mr Burnside yes[terday]?---Yes, I was trying to apply what my state of mind and what my thinking was then.
Hence, in answer to the question about which I am now asking you which was a weighing up of different risks and benefits you said that you would have had to have discussed it and that you would have sought advice from your doctor and relied on his advice?---Correct.
Continuity of the applicant’s use of Vioxx
742 The applicant said that, between May 2001 and September 2004, to the best of his recollection he took a Vioxx tablet every day. There may have been some days during this period when he did not take a tablet, for example, if he forgot (which he said would have been “very rare”) or if he had run out of tablets (which he also said occurred only rarely). If he omitted to take his tablets “for a day or two”, the pain would return and remind him to take Vioxx again. He used a seven‑day pill box, which he would take with him when travelling away from home and, when at home, which he would leave under his toothbrush or glasses in order to remind him to take his tablet each day.
743 Notwithstanding this evidence, the respondents submitted that I should find that the applicant failed to take Vioxx tablets for a period of about two months between the first week in October, and the first week in December 2003. That submission was based upon records obtained from Medicare of prescriptions obtained, and filled, under the PBS. It was the applicant’s consistent practice to obtain prescriptions and repeats from Dr Dickman (or from Dr Anderson − one of Dr Dickman’s colleagues at Tower Hill − on one occasion when Dr Dickman was not available). Relevantly to the present question at least, a prescription and three repeats were written each time, each providing for 30 days of medication. The records show that the applicant was prescribed a 30‑day course of Vioxx, and three 30‑day repeats, on each of 10 April 2002, 19 July 2002, 20 December 2002, 30 May 2003 and 2 December 2003. He had a prescription filled on or soon after each of those dates. He also had tablets dispensed under his repeats at approximately 30‑day intervals. Thus, after his prescription written (by Dr Anderson, as it happens) on 30 May 2003, the applicant was dispensed Vioxx on 1 June, 7 July, 3 August and 8 September 2003. The PBS records disclosed no prescription of Vioxx after that written on 30 May 2003 until 2 December 2003. If the records are complete and the applicant was acting consistently with what appeared to be his previous invariable practice, he would have visited Dr Dickman in about the first week of October 2003, to obtain a further prescription and repeats. There is no evidence that he did so. The respondents asked me to find that the applicant took no Vioxx tablets between his completion of the repeat which was dispensed on 8 September 2003, which would have occurred in early October 2003, and 2 December 2003, when the prescription which he obtained that day was filled.
744 According to the applicant, he became aware only in the course of preparation of the evidence for this proceeding that the PBS records showed the apparent gap in the dispensation of Vioxx tablets for him. Notwithstanding the PBS records, the applicant maintained his evidence that he took Vioxx every day. He could not explain the PBS records. While not objecting to the admission into evidence of the records as such, the applicant submitted that it could not be assumed that they accurately set out all of the prescriptions that had been written for him. It was submitted, correctly, that there was no evidence as to the nature or reliability of the systems used at the pharmacies to which the applicant had resort to record information about PBS prescriptions filled. Neither was there any evidence as to the nature or reliability of the systems used by Medicare to collect and record such information. It was submitted that some deficiency in the records themselves was at least as likely to be the explanation for the gap in the prescription of Vioxx which the records disclose. Although not stated in terms, the submission involves the proposition that I should find that the applicant did in fact obtain a prescription, and at least one repeat, in about the first week of October 2003. Previously, Dr Dickman’s practice had been to write a prescription with three repeats. Accepting the applicant’s submission would seem to involve the proposition that, on one occasion, Dr Dickman departed from his erstwhile practice, and gave the applicant one repeat only. I say that because a further prescription, with three repeats, was written for the applicant on 2 December 2003. What the applicant invites me to do is to base my inference upon the prospect that the PBS records, on this one occasion, failed to include any reference to a prescription which was in fact written, and to tablets which were in fact dispensed, and upon the further proposition that Dr Dickman’s previous, and subsequent, practice of giving the applicant three repeats was not followed. That is to say, in two separate and unrelated respects, the applicant asked me to base an inference upon a departure from normality. Unless other indications pointed quite strongly to the facts claimed to be inferred, I would be reluctant to do so.
745 It was put to the applicant that there was a good reason why he might have discontinued taking Vioxx in October 2003, namely, that it was at the start of that month that he gave up his employment with Teekay, thus removing the necessity for him to perform work which involved walking around ships, bending, climbing, getting into confined spaces and the like. The applicant rejected the suggestion that that circumstance made any difference to the regularity of his consumption of Vioxx. In closing submissions, his counsel pointed out that the PBS records showed that he continued to take Vioxx regularly after the beginning of December 2003, notwithstanding that, because of his heart attack, he was presumably taking things very easily at that time. As I shall demonstrate in a moment, however, if only the applicant’s PBS records are looked at, this submission is wrong in point of fact: after having had a prescription filled on 2 December 2003, the applicant next obtained Vioxx (on his own repeat) on 11 February 2004, an interval of 71 days.
746 That leaves the question: if the applicant took Vioxx in October and November 2003, how did he come by the tablets? After both the applicant and his wife, Mrs Julia Peterson, had given evidence, the applicant’s PBS records had been tendered and the applicant had been cross‑examined by reference to them, the PBS records which related to Mrs Peterson were tendered. Both she and her husband were then recalled for further cross‑examination. The focus of the debate then shifted to the question whether, if there were no Vioxx tablets available to the applicant after early October 2003 as a result of his own prescriptions and repeats, there were tablets available that had been prescribed for, but had not been taken by, his wife.
747 In her witness statement, Mrs Peterson had originally said that she took Vioxx once herself. She could not recall exactly when, but said that it would have been in about 2001 or 2002. Her doctor (not Dr Dickman or anyone from Tower Hill) prescribed Vioxx for her back pain, and gave her a sample pack. She thought that the sample pack was a week’s worth of tablets. However, medical and PBS records disclosed that she saw her doctor on 28 August 2003 with pain in the fifth finger on her right hand. Her doctor gave her a sample pack of Vioxx, which was 10 tablets. She accepted that she was mistaken to have supposed that her doctor prescribed Vioxx in about 2001 or 2002. Mrs Peterson saw her doctor again on 3 November 2003, because of a rash on her face and neck, which she was advised to treat with some cream available over the counter. At that time she obtained a prescription for Vioxx, with three repeats. She accepted that it was most likely that she had intimated to her doctor that the Vioxx which she had taken (the sample pack) had eased the pain in her finger, and that the doctor probably said, “well, I will give you a script for that medication”.
748 Mrs Peterson filled the prescription on the same day as she had obtained it, 3 November 2003. A repeat was filled on 23 November 2003. In each case, 30 tablets of Vioxx were dispensed. However, she rejected the suggestion that this sequence of events suggested that she then commenced to take the Vioxx for her own pain. She was firm in her denial that, although she had been prescribed a course of Vioxx, she never took any tablets. She said that this was because of a concern, which she first had about 30 years previously when speaking to another doctor, of becoming dependent upon medication. It was submitted on behalf of the applicant that I should accept Mrs Peterson’s evidence that, having obtained 30 tablets of Vioxx on 3 November 2003, she took none of them. If so, these 30 tablets would have been in the Peterson household at that time, and available to the applicant, had he run out of his own supply of Vioxx.
749 The respondents did not challenge the credibility of the applicant or his wife, save in the sense that they were mistaken as to the events of late 2003 of which they had little actual recollection. It was submitted that the most likely explanation for those events was that the applicant in fact took no Vioxx tablets between early October and early December (or at least early November) 2003. The respondents relied upon the fact that the explanation ultimately offered by the applicant was not originally advanced by him, before his wife’s PBS records for 2003 became available. They submitted that Mrs Peterson’s explanation was a most unlikely one. Her evidence involved the proposition that she had been given, and had taken, a sample pack of Vioxx tablets, and had found them effective in the relief of her pain; that she had returned to her doctor and obtained a prescription of Vioxx; that she had, on the same day, filled the prescription at a pharmacy; yet that she had failed to take any of the tablets so obtained, not through oversight or inadvertence, but by reason of a firm conviction that she ought not to take drugs because of the possibility of becoming dependent on them. The respondents also relied on Mrs Peterson’s evidence that she had never had a prescription in her own name filled with the intention of giving the tablets to her husband for him to take. Indeed, it was submitted on behalf of the respondents that neither the applicant nor his wife was the kind of person who would engage “in that sort of subterfuge and probably illegality without reason” and “there was no reason why they would engage in that sort of conduct”.
750 Mrs Peterson said that she had no specific recollection of her doctor having prescribed Vioxx on 3 November 2003, or thereabouts, but she was adamant that the only Vioxx tablets which she ever consumed were those from the sample pack. When the strangeness of her behaviour was put to her in cross‑examination, she said (speaking of the prescription written on 3 November 2003):
I would have perhaps filled it thinking that I would try them and then not gone on with taking the medication itself. I had the prescription, thought, ‘well, I will fill the prescription but whether or not I take it is up to me’, and I definitely did not take it.
This was, of course, ex post facto rationalisation on the part of Mrs Peterson, but it was not put to her that that explanation was a conscious untruth, or had been crafted purely for the purpose of supporting her husband’s claim. The sense I had of Mrs Peterson when she was giving this evidence was that she was conscientiously attempting to provide a viable explanation for what the PBS records showed, against her own clear recollection that she did not take any of the tablets which had been prescribed for her. I would also add that what might otherwise be viewed as the implausibility of Mrs Peterson’s evidence is rendered the less so when it is recognised that her appointment with her doctor on 3 November 2003 was not, apparently, for the purpose of obtaining Vioxx, nor even for purposes related to the pain in her finger. As noted above, she agreed with the inference proffered by counsel for the respondents that it was the doctor who had volunteered the proposal that more Vioxx should be prescribed. It is not the evidence that, on 3 November 2003, Mrs Peterson needed Vioxx for her own purposes.
751 To resolve these questions, I have found it useful to set out in a table the dates upon which the applicant had Vioxx prescribed for himself, the corresponding dates, commencing with 30 May 2003 (upon which a prescription was written), upon which he had each prescription, or a repeat, filled, the date of the 30th day after the date of each dispensation of Vioxx (ie the day upon which the applicant ought to have filled his next repeat, or new prescription, had he been taking Vioxx every day) and the numbers of days that he was, notionally, “late” in each case. I have prepared a second table, but this time including the dispensations of Vioxx done under the prescription written for Mrs Peterson on 3 November 2003.
752 The tables referred to are the following:
Timeliness of purchase of Vioxx – Applicant’s records only
|
Patient |
Date of Pres |
Date of Disp |
Prev +30 days |
Days “late” |
|
GP |
30-May-03 |
01-Jun-03 |
||
|
GP |
30-May-03 |
07-Jul-03 |
01-Jul-03 |
6 |
|
GP |
30-May-03 |
03-Aug-03 |
06-Aug-03 |
-3 |
|
GP |
30-May-03 |
08-Sep-03 |
02-Sep-03 |
6 |
|
GP |
02-Dec-03 |
02-Dec-03 |
08-Oct-03 |
55 |
|
GP |
02-Dec-03 |
11-Feb-04 |
01-Jan-04 |
41 |
|
GP |
02-Dec-03 |
15-Mar-04 |
12-Mar-04 |
3 |
|
GP |
02-Dec-03 |
08-Apr-04 |
14-Apr-04 |
-6 |
|
GP |
01-Jun-04 |
15-Jul-04 |
08-May-04 |
68 |
|
GP |
01-Jun-04 |
14-Aug-04 |
14-Aug-04 |
0 |
|
GP |
01-Jun-04 |
05-Sep-04 |
13-Sep-04 |
-8 |
Timeliness of purchase of Vioxx – Applicant’s & Mrs Peterson’s records
|
Patient |
Date of Pres |
Date of Disp |
Prev +30 days |
Days “late” |
|
GP |
30-May-03 |
01-Jun-03 |
||
|
GP |
30-May-03 |
07-Jul-03 |
01-Jul-03 |
6 |
|
GP |
30-May-03 |
03-Aug-03 |
06-Aug-03 |
-3 |
|
GP |
30-May-03 |
08-Sep-03 |
02-Sep-03 |
6 |
|
JP |
03-Nov-03 |
03-Nov-03 |
08-Oct-03 |
26 |
|
JP |
03-Nov-03 |
23-Nov-03 |
03-Dec-03 |
-10 |
|
GP |
02-Dec-03 |
02-Dec-03 |
23-Dec-03 |
-21 |
|
JP |
03-Nov-03 |
12-Jan-04 |
01-Jan-04 |
11 |
|
GP |
02-Dec-03 |
11-Feb-04 |
11-Feb-04 |
0 |
|
GP |
02-Dec-03 |
15-Mar-04 |
12-Mar-04 |
3 |
|
GP |
02-Dec-03 |
08-Apr-04 |
14-Apr-04 |
-6 |
|
JP |
03-Nov-03 |
13-May-04 |
08-May-04 |
5 |
|
GP |
01-Jun-04 |
15-Jul-04 |
12-Jun-04 |
33 |
|
GP |
01-Jun-04 |
14-Aug-04 |
14-Aug-04 |
0 |
|
GP |
01-Jun-04 |
05-Sep-04 |
13-Sep-04 |
-8 |
753 Looking at the first table, there were three occasions (shaded in the table) upon which the applicant would have been significantly late in obtaining further supplies of Vioxx if he were relying on his own prescriptions and repeats only: 2 December 2003, 11 February 2004 and 15 July 2004. However, if one assumes that he had available to him also his wife’s prescriptions and repeats, one of the delays (11 February 2004) is eliminated, and the others are about halved. It is not only the gap between 8 September 2003 and 2 December 2003 that may (in part) be accounted for by Mrs Peterson’s tablets: it is also the gap between 2 December 2003 and 11 February 2004 and that between 8 April 2004 and 15 July 2004. The apparent oddity that the applicant was – if he were taking his wife’s tablets – 21 days “early” when he obtained a prescription in his own name on 2 December 2003 is not seen to be so odd when it is realised that he visited Dr Dickman on that occasion not to obtain Vioxx but, as mentioned below, because of his chest pain.
754 The applicant and his wife frankly admitted that they had no recollection of the actual events with which I am here concerned. Mrs Peterson denied having obtained a prescription in her own name with a view to obtaining tablets for the applicant. For his part, the applicant denied “hoarding” (counsel for the respondents’ word) tablets. However, they both regularly took prescription drugs, and had the practice of keeping all the outstanding prescriptions and repeats together in a particular place. They thought it quite possible that the applicant (or his wife on his behalf) might have occasionally taken a prescription in Mrs Peterson’s name to the pharmacy with a view to obtaining Vioxx for himself. This was conjecture on their part, of course, but it strikes me as an explanation, as likely as any other, for the fact that repeats were filled on Mrs Peterson’s prescription on 23 November 2003, 12 January 2004 and 13 May 2004. It may be one thing to reject Mrs Peterson’s evidence that she did not take any of the tablets obtained by her on 3 November 2003, but it would be another thing altogether to find, in the face of her denials, that she consumed all of the tablets, the purchase of which is implied by those further repeats.
755 The applicant struck me as a credible witness who had a clear recollection of having followed a system which involved, more or less, the daily consumption of Vioxx. He was a systematic and careful man by nature and training, and brought to the taking of medication a degree of organisation which reflected that. He readily conceded that there were days when he omitted to take Vioxx, but I consider it unlikely that he would have allowed matters to lapse to the extent reflected in the first table set out above. A ready explanation for the gaps shown in that table not reflecting reality is provided by the pattern of dispensations under Mrs Peterson’s prescription. I am disposed to accept that explanation. Objectively, it introduces greater sense into what would otherwise be a less than credible pattern of consumption. An aspect of this conclusion is that I should find, as I do, that the tablets prescribed and dispensed on 3 November 2003 were most probably taken by the applicant rather than by his wife.
756 But the respondents next submitted that, on any view, there was a gap between about 8 October and 3 November 2003 in which the applicant had no Vioxx tablets to take, his own repeats having been exhausted and his wife’s prescription not yet having been written. It was suggested on behalf of the applicant that he might have been provided with a sample pack (or two) himself, but, given the previous regularity of prescriptions written for him, I think that unlikely. The applicant was not, at that time, in the position of someone contemplating taking a new drug. He had been on Vioxx for about 2½ years. I do not think there is any satisfactory explanation for how the applicant might have had Vioxx tablets available to him in this period. It would be speculation on my part to attempt to answer the question why the applicant did not visit his doctor and obtain another prescription (the only seemingly plausible explanation – that he had recently discontinued employment which required physical work – having been rejected by him). In the circumstances, I am not satisfied that the applicant had available to him more Vioxx tablets in the second half of 2003 than is implied by the second table above.
757 I should stress that the preparation of these tables had the purpose only of providing a base for the inference which I drew as to the applicant’s access to tablets obtained under his wife’s prescription. I do not infer that the actual daily pattern of consumption was as set out in the second table. I do not infer, for example, that the applicant took Vioxx religiously every day until 8 October 2003, after which there was a period of exactly 26 days when no tablets were taken. I think it much more likely that the days (occasional though they were) when the applicant overlooked taking his tablets were scattered randomly over the periods when he had tablets to take. Thus, for example, I think it likely that he did not “run out” of tablets on 8 October 2003 exactly, but had some tablets which probably carried him into the next few days.
758 On any view, however, there was a period of three weeks or thereabouts leading up to 3 November 2003 when the applicant did not take Vioxx. I do not accept his evidence to the contrary. I am not, however, disposed to use that circumstance to qualify my acceptance of the applicant’s evidence that, generally, he took Vioxx every day. Specifically, I find that the applicant took Vioxx between 3 November 2003 and his heart attack about five weeks later.
The applicant’s heart attack
759 In early December 2003, the applicant began to experience an ache or pain in his chest, which would last for 10‑15 minutes and then go away. On 2 December 2003, he saw Dr Dickman, who noted that the applicant’s blood pressure was 155/90. Dr Dickman’s working diagnosis was reflux oesophagitis and hypertension. Dr Dickman was concerned that the applicant’s chest pain may have been angina, so he prescribed Atenolol, a drug to lower blood pressure and to stabilise any cardiac rhythm disturbance. Various pathology tests were arranged, including an electrocardiogram (“ECG”) which was done on 3 December 2003. After the ECG test, the nurse told the applicant that he could go, which he took as an indication that his results were normal. As it happened, however, the ECG showed T‑wave inversion in leads I and aVL with a comment that this was possibly due to sub‑endocardial ischaemia or left heart strain. The cardiologists agreed that the applicant “definitely had obstructive atherosclerotic coronary disease with an acute coronary syndrome” at this time. Profs Harper and Celermajer added, in their oral evidence (the latter noting that all the cardiologists were agreed on the point), that what caused the applicant’s pain in early December 2003 was plaque rupture. However, the applicant did not know of the ECG results.
760 On 4 December 2003, the applicant drove to Canberra with his wife for a work commitment. Dr Dickman telephoned him on that day (inferentially to inform him of the results of the ECG), but the applicant missed the call. After completing his commitment in Canberra, the applicant and his wife continued on to NSW for a friend’s birthday. They returned to Melbourne on Sunday 7 December 2003. On the morning of that day, the applicant experienced another episode of chest pain for about ten minutes. Mrs Peterson gave evidence that her husband was not feeling well, as a result of which she drove the car. They arrived home at about 9.30 or 10.00 pm and retired.
761 In the early hours of 8 December 2003, the applicant experienced another episode of chest pain which was worse than the previous ones. The pain was severe, and radiated down both his arms. Soon after it began, he started losing his peripheral vision. He felt as though he had been hit very hard in the solar plexus. He said he was “stunned and almost choking, and couldn’t regain my breath no matter how hard I tried”. He was doubled over and gasping. He described the pain as “a crushing type of pain, like I was suffering a massive cramp across all of my upper body”. Realising that something was very seriously wrong, the applicant took some aspirin (this was at about 1.00 am) and had a lukewarm shower. Then he and his wife went to the Frankston Hospital, where he arrived at about 3.00 am.
762 What occurred thereafter, as derived from hospital records, was described by Prof Vaughan as follows:
[The applicant] was found to have acute electrocardiographic changes including ST elevation in multiple contiguous leads and was diagnosed with an anterior myocardial infarction. … He was admitted and treated with nitroglycerine and thrombolytic therapy. … His initial cardiac enzymes drawn at 3.47 am were not elevated; however, by 7.00 am he had elevated markers of myocardial injury, including elevated troponin and CK‑MB.
[The applicant] underwent cardiac catheterization on December 11, 2003. At the time of coronary angiography, [he] was found to have significant coronary artery disease, including a 90% stenosis in his proximal left anterior descending artery (LAD), a tight ostial stenosis in his LADD1 (first diagonal artery off of the LAD), and minor disease in his right coronary artery (RCA). … His left main and left circumflex arteries were noted to be free of disease. … His ejection fraction was calculated at 40%. … He underwent successful PTCA and stenting to his proximal LAD without complication. … Final angiographic result was noted to be excellent with 0% residual stenosis.
Prof Harper gave evidence to much the same effect.
763 Prof Harper examined the applicant in October 2008. He gave evidence that, since the applicant’s heart attack, he has been given lifestyle modification and has been treated with a standard cardioprotective pharmacological regime. As part of the cardiac catheterisation which the applicant underwent on 11 December 2003 he had a left ventriculogram which showed antero-apical and apical wall hypokinesia. This meant there was reduced contraction as a result of the heart attack in a large portion of the left ventricle. The ejection fraction – a measure of overall cardiac strength – was 40%. A normal ejection fraction is greater than 50%. Prof Harper said that, when heart muscle is damaged by a heart attack, the ejection fraction does not necessarily fall unless the extent of damage has been large. This was because the undamaged heart muscle would compensate for the damaged muscle by contracting more strongly. The fact that the applicant’s ejection fraction was significantly below normal indicated that he had had a large heart attack. Subsequent echocardiograms confirmed that the applicant’s heart attack was a large one.
Did Vioxx cause or contribute to the applicant’s heart attack?
764 The cardiologists said that the applicant’s risk factors for myocardial infarction in 2001 were hypertension, hyperlipidemia, obesity and the presence of left ventricular hypertrophy. He was, the cardiologists agreed, “highly likely to have had coronary atherosclerosis that was clinically silent at the time”. In addition, he was a former smoker, of male gender and aged 51 years. He was at moderate to high risk of a cardiovascular event over the next 5 years. Prof Harper estimated that risk at 20‑25%, and the other cardiologists took no issue with that.
765 Putting aside the possibility of the consumption of Vioxx as such increasing the risk, the cardiologists agreed that, by December 2003, the applicant’s risk factors were “slightly improved”. They noted that the applicant had lost weight and that his lipid profile had improved, although still abnormal. They considered that the other risk factors were essentially unchanged. The cardiologists were unable to say whether the consumption of Vioxx had affected the applicant’s blood pressure, as there were insufficient data (only three blood pressure readings having been taken since the applicant commenced to take Vioxx). I should add that, although counsel for the applicant made the general submission that the consumption of Vioxx did lead to elevated blood pressure (a subject which I addressed at paras 532‑535 above), they did not submit that this occurred in the case of the applicant himself. In any event, the evidence would be insufficient to sustain any such finding. Neither, in the applicant’s case, was there any attention given to the question whether it was elevated blood pressure that led to his atherosclerosis as it existed in December 2003, and thus indirectly contributed to his heart attack.
766 The cardiologists were agreed that, in December 2003, the applicant had a large acute anterior ST segment elevation myocardial infarction. The aetiology of that heart attack was thrombosis consequent upon a ruptured atherosclerotic plaque in his proximal left arterial descending coronary artery. In terms which, while not referred to by the other cardiologists, were not the subject of any challenge by the respondents, Prof Harper explained what most likely happened to the applicant.
The … findings at coronary angiography are consistent with the hypothesis that Mr Peterson had developed a significant athero-sclerotic plaque in his left anterior descending artery on which a thrombus, or blood clot, formed, thus completely obstructing the artery producing a ST segment elevation myocardial infarction. Most likely the surface of the athero-sclerotic plaque had ulcerated in the week prior to the actual heart attack, causing intermittent thrombus formation and partial obstruction resulting in the paroxysmal episodes of chest pain Mr Peterson experienced. Eventually the thrombus completely obstructed the artery, thus producing the anterior ST segment elevation infarction on 8 December 2003. The thrombolytic therapy that he was given that day presumably dissolved the clot leaving the residual atherosclerotic plaque, which was subsequently treated by angioplasty and stenting on 11 December 2003.
767 The more difficult part of the task confronting the cardiologists was to form a view, so far as possible, about the contribution of Vioxx to the events in the vasculature as described by Prof Harper. They agreed that, in a patient with multiple risk factors such as the applicant, there was no way definitively to determine which risk factor caused the heart attack. Neither was there any specific feature of the applicant’s condition that established whether Vioxx did, or did not, cause or contribute to the heart attack. Prof Celermajer put the matter as follows:
There are many 53 year old men with Mr Peterson’s risk factor profile who have heart attacks around the world. You wouldn’t be staggered at the situation that absent Vioxx Mr Peterson would have had a heart attack and as my learned colleagues have pointed out, there’s no footprint of Vioxx, there is no signature of Vioxx to indicate that a particular heart attack was a Vioxx heart attack versus a common garden variety heart attack you have every day.
That is to say, the applicant may have had the very heart attack which he did have, at the same time and of the same level of seriousness, had he not been taking Vioxx.
768 In this state of things, it is perhaps unsurprising that the cardiologists were not agreed on the question whether Vioxx contributed to the applicant’s heart attack. Earlier in these reasons, I have set out their respective views on the general question whether Vioxx increased the risk of CVT events, and of myocardial infarctions, occurring. As to the applicant specifically, Profs Zipes and Harper expressed the view that the consumption of Vioxx increased the risk of him having his heart attack by a factor of about two. They were asked whether that was the same thing as saying that they believed it twice as likely that the applicant’s consumption of Vioxx in fact caused, or in some material way contributed to, the heart attack which in fact took place. Prof Zipes answered as follows:
That number comes primarily from the APPROVe trial where the relative risk was around 2. When an individual has multiple risk factors and suffers a myocardial infarction it’s very difficult to say one specific risk factor was responsible for the infarct versus the constellation of risk factors all interacting and that summation producing the infarct. I think more likely than not had he not been taking Vioxx he would not have had an infarct at that time but it’s difficult to say that Vioxx was the specific cause of that infarct. I think we are dealing with statistics and the likelihood of him having an infarct is increased by him taking Vioxx, so I think to a reasonable degree of medical probability Vioxx played a substantial contributing role.
Prof Harper reiterated that, in his view, Vioxx doubled the applicant’s risk of having a heart attack. When asked to look at the matter ex‑post and to express a view as to whether Vioxx in fact contributed to the applicant’s heart attack, Prof Harper said that it was a difficult question, “but if it is more than two times then it’s more likely than not that Vioxx contributed to the heart attack”. Later in his evidence, Prof Harper revised upwards his estimate of the extent to which Vioxx increased the applicant’s risk of heart attack. Looking at the data from APPROVe, he said:
There were 21 myocardial infarctions in the rofecoxib group and nine in the placebo group; slightly more than 2:1. That is the data that I would rely on over and above any other data that’s been presented.
769 When Prof Harper said that a risk of “more than two times” meant that it was more likely than not that Vioxx contributed to the applicant’s heart attack, he was implicitly invoking a rule of thumb that says that if, for example, in two otherwise identical groups of people, the events of interest are twice as numerous in the group taking a particular drug as in the placebo group, we may infer that half of those in the former group who experienced an event would have done so anyway. From this it is said that a relative risk of 2.0 goes no further than to imply that an event which occurred in the drug‑taking group is equally likely to have done so as a result of the work of the drug as it is to have occurred in any event. This subject is explained in the judgment of Spigelman CJ in Seltsam Pty Ltd v McGuiness (2000) 49 NSWLR 262, 280‑285. Concluding, his Honour said (at 285 [137]):
In Australian law, the test of actual persuasion does not require epidemiological studies to reach the level of a relative risk of 2.0, even where that is the only evidence available to a court. Nevertheless, the closer the ratio approaches 2.0, the greater the significance that can be attached to the studies for the purposes of drawing an inference of causation in an individual case. The “strands in the cable” must be capable of bearing the weight of the ultimate inference.
770 The attribution of a contributory role to Vioxx in the applicant’s heart attack, then, came down to an interpretation of the risk data yielded by the various trials and studies and to an opinion as to the relevance of those data to the circumstances of the applicant particularly. Inescapably, this required the cardiologists to form a view as to the validity of the FitzGerald hypothesis. Despite what appeared to be a certain agnosticism about the hypothesis, Prof Harper expressed the opinion that the observed riskiness of Vioxx was due to its effect in the post‑plaque rupture vasculature. This was, I consider, an implicit endorsement of FitzGerald. He and Prof Zipes considered that the applicant’s consumption of Vioxx increased his risk of having a heart attack by approximately a factor of two (which they derived from APPROVe). Profs Celermajer and Vaughan did not agree. They expressed their conclusion thus: “We do not believe, with a reasonable degree of scientific certainty, that Vioxx played a role in causing [the applicant’s] heart attack.” When asked by the court what was meant by “reasonable degree of scientific certainty”, Prof Celermajer said:
We were presented with a binary outcome – it contributed or it didn’t. We were not invited to say with what degree of possibility might it have contributed. So when asked to make a binary outcome decision we responded to that by saying we did not. We simply wished to acknowledge that we are not absolutely certain of that because the data themselves are [uncertain] ….
771 Unlike Prof Celermajer, I am required to make an assessment of the probability that Vioxx caused or contributed to the applicant’s heart attack. Here the actual circumstances of the applicant on 8 December 2003 are important. I accept the evidence of Prof Vaughan that Vioxx did not cause atherosclerosis and did not cause plaque rupture. However, the cardiologists are agreed that the applicant had clinically silent atherosclerosis in and subsequent to 2001, and that he experienced plaque rupture in early December 2003. It seems highly probable that this would have been a clinical diagnosis positively made had the applicant’s interstate trip not caused him to miss Dr Dickman’s phone call. However that may be, the fact is that, after plaque rupture, the applicant was in the high risk group referred to in the APPROVe results – he had symptomatic atherosclerotic cardiovascular disease. He may have been in that situation earlier, but on 2 December 2003 he unquestionably was.
772 As the respondents and their experts pointed out, if valid, the FitzGerald hypothesis proposes an acute risk of thrombotic outcomes from the consumption of Vioxx. That there should be such a risk, however, requires first that there be a potentially thrombotic environment in the vasculature. That environment was, in the applicant’s case, present in December 2003. I think it likely that the applicant’s was a case in which the absence of prostacyclin in the immediate vicinity of his injured artery made some contribution to the formation of the thrombus or thrombi that caused the occlusion which gave him his heart attack. I do not find that the heart attack would not otherwise have occurred: there is no way of knowing whether it would have. But I am satisfied that some material contribution was played by the brake on the synthesis of prostacyclin applied by the inhibition of COX-2, and therefore by the consumption of Vioxx.
773 It is a necessary premise of this finding that the applicant was taking Vioxx, more or less daily, in the weeks leading immediately to early December 2003. As I have found earlier, he was doing so because he had access to Vioxx obtained under his wife’s prescription. Had that not been the case, I would not have been able to find, on the probabilities, that Vioxx made a material contribution to the applicant’s heart attack. Here I refer to my treatment of the cardiologists’ evidence as to the relevance of the pattern of use of Vioxx at paras 548‑550 above. While I recognise the force of Prof Harper’s evidence as set out in para 519 (which provided a measure of support for Prof Zipes’ more categorical evidence on the same subject), in the light of my reasons on mechanism, and given the absence of any trial data with respect to risk in a situation of discontinuous administration of the drug, I could not be satisfied that Vioxx would, merely by having been taken by the applicant continuously down to about (say) the middle of October 2003, have contributed to what happened on 8 December 2003. In the words of Spigelman CJ in Seltsam, there would then have been insufficient “strands in the cable” to support such a finding.
Consequences of the applicant’s heart attack
774 I have referred (at para 763 above) to the evidence given by Prof Harper as to the damage done to the applicant’s heart muscle as a result of his myocardial infarction on 8 December 2003. Prof Harper said that this damage was permanent. He said that, at present, the applicant has only mild overall impairment of heart function, but he has a significantly diminished cardiac reserve. If he were to suffer a further heart attack and sustain further damage to his heart, his ejection fraction would fall further, putting him at significant risk of developing cardiac failure and increasing his susceptibility to death.
775 Prof Harper referred also to a number of matters of which he was apparently informed by the applicant, namely, that he continues to work as a self-employed marine consultant but by choice does less work than previously; that he avoids locations which lack immediate high quality cardiac care if needed; that he no longer scuba dives or undertakes heavy physical activity; that he tires more easily; and that he feels short of breath on exertion much more readily than prior to his heart attack. The applicant informed Prof Harper that he experienced intermittent episodes of chest pain. In Prof Harper’s view, these were not necessarily related to exertion. A negative result to an exercise test recently undertaken by the applicant indicated to Prof Harper that the chest pain was not due to recurrent myocardial ischaemia, but was probably anxiety-related; nonetheless it caused the applicant great concern. Prof Harper said that the applicant consistently had symptoms which corresponded to activities which required 5-6 METS of energy. Taking these physical and psychological factors into account, Prof Harper considered that the applicant’s level of “whole person impairment” as a result of his heart attack was in the order of 25-30%.
776 In Prof Harper’s view, the applicant’s current risk of having a further heart attack has been significantly reduced by the facts that he is no longer taking Vioxx, that he has an improved lifestyle and that he is currently on a strong pharmacological cardioprotective regime. Prof Harper estimates the applicant’s current risk of suffering a further heart attack in the next five years to be in the order of 5-10%.
777 As a result of the applicant’s heart attack, he was not able to continue with so much of his plans as involved GPA providing safety consultancy services directly to industry. Instead, through GPA, he has undertaken work for another safety consultancy company, Safety Wise Solutions Pty Ltd (“SWS”). There he does the same kind of work as he would have done had GPA provided such services directly, that is, he runs training courses, he undertakes safety audits, he performs safety inspections and he carries out accident and incident investigations. He also does some work as a consultant for P & O Maritime Services.
778 In the small team of consultants used by SWS, the applicant is the only one with substantial experience in the maritime industry. The managing director of SWS, Mr Gerry Gibb, holds the applicant in high regard. In the kind of work that the applicant does, it is often a requirement to be on a site for ten or more consecutive days before taking a break. However, the applicant finds that he is able to work for about six days straight at most. He tends to get too tired to work much more than that. The applicant told Mr Gibb that he did not really want to work more than 8-10 days each month. According to Mr Gibb, if the applicant wanted to work more, he could have as much work as he wanted. SWS is always very busy, and always has work available for the applicant.
779 Before he allocates work to the applicant, Mr Gibb assesses whether the particular assignment would be suitable for the applicant, having regard to his heart condition. About 20% of the applicant’s work is undertaken outside Australia, in relation to which Mr Gibb considers the availability and standard of medical facilities at the location in question, and the length and arduousness of the travel required to get there. Thus, for example, Mr Gibb perceived that there would be no difficulty with the applicant being sent to Dubai, where the medical facilities are excellent and the site in question was reasonably accessible, but chose not to send the applicant to a particular job in South Africa because travel to the site required a very long journey by car. This latter consideration is also taken into account by Mr Gibb when allocating work to Mr Peterson to remote sites within Australia. I rather gather from Mr Gibb’s evidence, however, that it is the applicant’s self-imposed limitation of 8 − 10 days per month, rather than any of these considerations, that ultimately provides the practical restraint upon the amount of work that he performs for SWS.
780 The applicant did not suggest that the operation of his “Lockout” business had been affected by heart attack. Indeed, in the financial year to 30 June 2008, sales from that business (which is also conducted through GPA) exceeded $550,000.
781 Recreationally, the applicant’s heart attack has obliged him to give up diving, which he used to enjoy.
part vii − the applicant’s case in negligence
The existence and content of a duty of care
782 The applicant submitted that “[a] manufacturer of a product intended for human consumption owes a duty to intended ultimate consumers to take reasonable care to prevent the product from causing injury or loss to those consumers …”. At the general level, the respondents’ articulation of this proposition was, if anything, even broader. They referred to the principle as stated by McHugh J in Dovuro Pty Ltd v Wilkins (2003) 215 CLR 317, 328 [29]:
It is beyond doubt that a manufacturer of any product owes a duty to a consumer to take reasonable care to prevent the product causing injury or loss to the consumer.
His Honour said much the same thing in Graham Barclay Oysters Pty Ltd v Ryan (2002) 211 CLR 540, 585 [106]. The respondents added − and I have little doubt but that the applicant would agree − that “[a] manufacturer’s duty includes responsibility for ensuring that appropriate and necessary information about the product is communicated to persons who will use or consume the product, and who it can be foreseen may suffer loss or damage …”.
783 In the applicant’s written submissions, it was contended that:
c. The scope and content of the duty of care owed by the manufacturer of a product to a consumer will be limited to taking reasonable steps to avoid injuries which are reasonably foreseeable …
d. The particular injury suffered need not be foreseeable. It is sufficient if the injury suffered is of a general type or kind which is foreseeable …
e. A risk of injury is foreseeable if it is real and not far‑fetched or fanciful, even if remote and extremely unlikely to occur …
f. In order for a duty to extend to a particular risk, it must be one that is “not insignificant”: Wrongs Act 1958 (Vic), s 48(1)(b). “Insignificant risk” includes risk which is far‑fetched or fanciful, and a “not insignificant risk” is a risk other than an insignificant risk, and includes a significant risk: Wrongs Act 1958 (Vic), s 48(1)(b).
The first three of these propositions were supported by references to authority. All four of them are uncontroversial at the general level. Only in one respect did the respondents take issue with the applicant’s articulation of the law in these terms. They made the following submission:
[T]he Respondents’ duty must be informed by, and not inconsistent with, the peculiar regulatory regimes which regulated all of their actions in bringing Vioxx to market, and also in light of the fact that Vioxx could only be obtained on prescription from an appropriately qualified healthcare professional. … Accordingly, … any such duty is discharged by the provision of information to the TGA in accordance with the applicable legislation and in satisfaction of the TGA’s requirements and the provision of information to prescribers in a way which is limited by and complies with the relevant legislation and the conditions of the medicine’s registration on the ARTG.
It will be apparent, however, that this submission is essentially concerned not with the content of the duty of care, but with its discharge. Indeed, in a passage in Ryan immediately following that on which the respondents relied, McHugh J said:
To formulate the duty in more specific terms invites error because it is likely to mix a question of law (whether a duty existed) with a question of fact (whether a breach occurred). If the duty is formulated in specific terms, the issue on breach is whether the duty has been performed in accordance with the terms of the duty as formulated. But, as Wyong Shire Council v Shirt ([146 CLR 40 at 47‑48, per Mason J, Stephen and Aickin JJ agreeing]) shows, the question of breach is far more complex than an affirmative or negative answer to the question whether the defendant carried out the duty as formulated. It involves evaluating and weighing a number of competing considerations.
I shall deal with the respondents’ point as to the sufficiency of their compliance with the regulatory regime for which the TG Act provides below, as part of my consideration of the steps which MSDA took in the discharge of the duty of care.
784 The applicant also submitted as follows:
A duty of care will only arise in respect of risks that are “material”. A risk will be “material” if a reasonable person in the consumer’s position would be likely to attach significance to it or the manufacturer was or should have been aware that the consumer would be likely to attach significance to it: see Rosenberg v Percival (2001) 205 CLR 434 at [75]; Rogers v Whitaker (1992) 175 CLR 479 at 490 [16]. In order to satisfy the “materiality” requirement it is not necessary to establish that the patient would have acted upon a warning if given – this is a question of causation. The test is “somewhat lower than that” and materiality will be established if the reasonable or particular person “would have been likely seriously to consider and weigh up the risk before reaching a decision”: Rosenberg v Percival (2001) 205 CLR 434 at [79], [80].
Rogers v Whitaker was a medical negligence case. In the course of their joint reasons, Mason CJ, Brennan, Dawson, Toohey and McHugh JJ considered the judgment of King CJ in F v R (1983) 33 SASR 189, noting that his Honour had been of the view that −
… the amount of information or advice which a careful and responsible doctor would disclose depended upon a complex of factors: the nature of the matter to be disclosed; the nature of the treatment; the desire of the patient for information; the temperament and health of the patient; and the general surrounding circumstances.
(175 CLR at 488). In a paragraph which contains a passage relied on by the respondents in the present case, their Honours in Rogers v Whitaker said (175 CLR at 490):
We agree that the factors referred to in F. v R.. by King C.J…. must all be considered by a medical practitioner in deciding whether to disclose or advise of some risk in a proposed procedure. The law should recognize that a doctor has a duty to warn a patient of a material risk inherent in the proposed treatment; a risk is material if, in the circumstances of the particular case, a reasonable person in the patient’s position, if warned of the risk, would be likely to attach significance to it or if the medical practitioner is or should reasonably be aware that the particular patient, if warned of the risk, would be likely to attach significance to it. This duty is subject to the therapeutic privilege.
This appears to be the supposed jurisprudential basis for the applicant’s submission that a duty of care will only arise in respect of risks that are “material”.
785 In Rogers v Whitaker, however, the existence of a duty of care was not controversial: see 175 CLR at 483. Neither was it controversial in Rosenberg v Percival. The test of materiality was formulated (in both cases) in the context of the question whether the discharge of the duty of care required a medical practitioner to warn his or her patient of risks − even very unlikely risks − known to be involved in a procedure under contemplation. In such a context, that test has, if I may so observe with respect, a particular relevance, as it provides a framework for applying the general law of negligence to the circumstances of a doctor‑patient relationship where the doctor has a duty to advise but the patient has the ultimate decision.
786 By contrast, the present is a product liability case. The notion of materiality finds no express mention in the classic formulation of Lord Atkin in Donoghue v Stevenson [1932] AC 562, 599 that −
… a manufacturer of products, which he sells in such a form as to show that he intends them to reach the ultimate consumer in the form in which they left him with no reasonable possibility of intermediate examination, and with the knowledge that the absence of reasonable care in the preparation or putting up of the products will result in an injury to the consumer’s life or property, owes a duty to the consumer to take that reasonable care.
Indeed, as noted above, the parties in the present case accepted such a proposition. I consider, in the circumstances, that the applicant was wrong to submit that a duty of care arises only in respects of risks which are “material”. In my view, on the facts of the present case, the duty of care was a general and universal one arising from the proximity between the respondents and all who fell within the class of persons for whom Vioxx was intended − and to whom it was conventionally prescribed − as a packaged product for human consumption.
787 Before completing this section of my reasons, I should refer to the potential relevance of one aspect of the applicant’s evidentiary case to the question whether MSDA owed a duty of care to him. I have found that the Vioxx tablets which the applicant consumed between 3 November and 2 December 2003 had been, as a matter of probability, obtained under a prescription written for his wife. The respondents did not submit that, should I make a finding in those terms, they did not owe the applicant a duty of care, or their duty of care was somehow qualified, by reason of that circumstance. Subject to their other points to which I refer, the respondents approached the matter as though the applicant were within the class of persons to whom they owed the duty.
Discharge of the duty of care
788 In the course of their written submissions, the respondents advanced three general propositions on the subject of the discharge of their duty of care. It is convenient to deal with them at the outset.
789 The first proposition was that compliance with the requirements of the regulatory regime for which the TG Act provided constituted, as a matter of law, a sufficient discharge of the duty of care which fell upon the manufacturer of prescription medicines such as MSDA. The respondents submitted that the TGA was the “independent expert evaluator of prescription medicines” and that –
(a) one of the means by which the TG Act does this is a system of registration and listing of medicines which requires the TGA to be satisfied of the quality, efficacy and safety of a prescription medicine for its intended purposes before that medicine can be marketed in Australia. The TGA also has the power (delegated to it by the Secretary of the Department of Health & Ageing) to impose conditions on its approval of a medicine: ss 25(1) and 28 of the TG Act;
(b) the TGA’s review of a medicine involves a detailed consideration of the data submitted by the sponsor (in this case MSDA), including review by expert evaluators and consideration by a committee of expert advisors (ADEC);
(c) TGA review continues throughout the life of the medicine;
(d) in the case of Vioxx, the TGA reviewed three Category 1 applications: the original application for approval, the application to incorporate the VIGOR data into the PI, and the application for approval of an indication for symptomatic treatment of rheumatoid arthritis;
(e) in addition, the TGA received Periodic Safety Update Reports from MSDA throughout the time Vioxx was on the market.
790 The respondents submitted that the system of evaluation of prescription medicines established by the TG Act was comprehensive. Implicit in it was a recognition that prescription medicines carried with them inherent risks. The system struck a balance between the need to protect the public from the harmful, and the public’s need for the beneficial, effects of medicines. The Act did this by providing for a detailed review regime, and “a system for the provision of information and warnings to doctors in a prescribed form”. The regime sought to strike a balance between the need to test prescription medicines before they were marketed so as to understand their risks and the interest of the public in obtaining access to medicines sooner rather than later. The respondents submitted that it would be inconsistent for the law of negligence to impose a duty of care upon manufacturers of prescription pharmaceuticals beyond the duty to participate in this regulatory scheme in a bona fide manner, and to fulfil their statutory obligations under the scheme.
791 The objects of the TG Act were set out in s 4(1) as follows:
The objects of this Act are to do the following, so far as the Constitution permits:
(a) provide for the establishment and maintenance of a national system of controls relating to the quality, safety, efficacy and timely availability of therapeutic goods that are:
(i) used in Australia, whether produced in Australia or elsewhere; or
(ii) exported from Australia;
(b) to provide a framework for the States and Territories to adopt a uniform approach to control the availability and accessibility, and ensure the safe handling, of poisons in Australia.
Consistently with these objects, the system established by the TG Act was essentially a regulatory one, such as legislation might regulate, for example, the driving of cars on public roads, the standards of construction of new buildings or the guarding of dangerous machines. There was nothing that I could find in the TG Act – and there was no provision to which the respondents drew my attention – that purported to confine what would otherwise be required in the reasonable discharge of the duty of care owed by a drug manufacturer to those who consumed it. Notwithstanding the absence of any such provision, was it a matter of necessary implication that the TG Act operated in the way proposed by the respondents?
792 The respondents’ submissions fell a good distance short of persuading me that any such implication should be made. They contained no such rigorous examination of the terms of the TG Act as would be necessary to make good the point. In one respect at least, the submissions overreached the actual terms of the Act. I refer to the proposition that the TG Act provided for “a system for the provision of information and warnings to doctors in a prescribed form”. The respondents did not identify where the Act so provided. For my own part, I have been able to find nothing in the TG Act which either mandated or regulated the terms in which a drug manufacturer such as MSDA provided information and warnings to doctors.
793 The effect of accepting the respondents’ submission would be that, so long as the manufacturer of a prescription medicine complied in good faith with the system for which the TGA provided, it could never be held to have fallen short in the discharge of its duty of care to consumers. The manufacturer, could, it seems, engage in all manner of negligent promotion, communication or presentation without being exposed to claims of the kind that the applicant now brings. A careless error in the preparation of a particular production batch (ie a snail‑in‑the‑bottle problem) could never be held to constitute a failure to discharge the duty of care. I highlight here, of course, the circumstance that the respondents’ point fails to address quite egregious negligent acts of commission. I do not know how the respondents might respond to these criticisms: their written submissions proceeded more by way of manifesto than by way of analysis. They were not developed orally.
794 By placing on the market a product to be consumed by end users, the manufacturer of a prescription medicine, no less than the manufacturer of any other product intended for human consumption, establishes the setting for the creation of the relationship of proximity from which the common law duty of care arises. I would be slow to hold that a law which did not in terms deal with that very subject implicitly qualified the manufacturer’s obligation – as it would otherwise be defined by the common law – so as to permit it, in effect, to act less than reasonably in the discharge of the duty, and to act in a way that produced loss or damage, yet to be shielded from claims by injured parties. There is, in my view, nothing unworkable or anomalous about such a manufacturer remaining under an obligation to take reasonable steps to avoid loss or injury to the end user at the same time as being required to comply with the regulatory system for which the TGA provided. The manufacturer’s obligation is not, in my view, exhausted upon compliance with the statute – no more so than the motorist’s obligation to take care in the driving of his or her vehicle is exhausted upon compliance with road traffic regulations: see Sibley v Kais (1967) 118 CLR 424, 427.
795 For the above reasons, I reject the respondents’ point that the steps required in the discharge of their duty of care went no further, as a matter of law, than to participate in the system of regulation for which the TG Act provided. That conclusion does not, of a course, exclude the prospect that, in identifying the steps required in the discharge of the respondents’ duty of care, the requirements of that system, and MSDA’s compliance with them, might not be relevant under the general law. To the extent relevant, this is a matter to which I shall turn in due course.
796 The respondents’ second general proposition was that the court should recognise the existence of the “learned intermediary” defence which is available in many jurisdictions in the United States in product liability proceedings involving prescription drugs. The nature of the defence was explained by the Fifth Circuit Court of Appeals in Reyes v Wyeth Laboratories 498 F 2d 1264 (1974) at 1276:
We cannot quarrel with the general proposition that where prescription drugs are concerned, the manufacturer’s duty to warn is limited to an obligation to advise the prescribing physician of any potential dangers that may result from the drug’s use. This special standard for prescription drugs is an understandable exception to the Restatement’s general rule that one who markets goods must warn foreseeable ultimate users of dangers inherent in his products. See Restatement (Second) of Torts, Section 388 (1965). Prescription drugs are likely to be complex medicines, esoteric in formula and varied in effect. As a medical expert, the prescribing physician can take into account the propensities of the drug as well as the susceptibilities of his patient. His is the task of weighing the benefits of any medication against its potential dangers. The choice he makes is an informed one, and individualized medical judgment bottomed on a knowledge of both patient and palliative. Pharmaceutical companies then, who must warn ultimate purchasers of dangers inherent in patent drugs sold over the counter, in selling prescription drugs are required to warn only the prescribing physician, who acts as a “learned intermediary” between manufacturer and consumer.
The defence was referred to by Kiefel J in Carey‑Hazell v Getz Bros & Co (Aust) Pty Ltd [2004] FCA 853 at [219]. In a passage relied on by the respondents, her Honour said:
The effect of a “learned intermediary” upon a manufacturer’s duty has been the subject of considerable case law in the United States. In Sterling Drug, Inc. v Cornish 370 F.2d 82 (8th Cir 1966) the term was used in relation to a physician who acted as a liaison between patient and drug manufacturer. The content of the manufacturer’s duty was to warn prescribing physicians. … It has been explained that the reason for describing the duty in this way is because it is a physician’s duty to remain abreast of product characteristics and to decide which facts should be told to the patient. Once adequate warnings are given to the physician, the choice of treatment and the duty to disclose properly fall upon the doctor.
With respect, as I understand the defence, it is not based on the assumption that a physician will “remain abreast of product characteristics”. Quite the contrary: the duty to warn the physician seems clearly to be based on the silent proposition that, absent warnings, the physician may know little about the characteristics of a product alleged to be harmful. However, the defence does involve the assumption that a physician, with his or her training and understanding, will be best equipped to receive a warning from the drug supplier (which may well be quite technical and unintelligible to many patients) and to factor that into the many considerations taken into account when making prescribing decisions.
797 The learned intermediary defence was not resorted to by the court in Carey‑Hazell, and has not been the subject of any authoritative judgment in Australia. I consider that, expressed as it was in the extract from Reyes set out above, it may be too categorical an articulation of the duty of a manufacturer of a product intended for human consumption. As so expressed, the test almost defies one to contemplate situations in which the broad duty deriving from Donoghue v Stevenson may not be sufficiently discharged, even in cases of prescription medicines, by warnings given to physicians. There may, for example, be unusual lifestyle choices made in the future by the consumer which, being beyond the range of matters reasonably inquired about by the physician, might later lead to adverse reactions. However, nothing further needs to be said about such speculations, or about the defence generally, since I consider that the present case can and should be decided without reference to the learned intermediary defence as such. As will appear, I consider that the general principles adumbrated in the cases to which I have referred provide a sufficient framework for the making of the judgments necessary to resolve the issues presented here.
798 The respondents’ third general proposition was that any identification of the steps required of them in the discharge of their duty of care had to proceed on a case‑by‑case basis and could not be done generally, that is to say, so as to produce an answer that was applicable to all group members. They founded the point upon the applicant’s allegation in his Further Amended Statement of Claim that the consumption of Vioxx materially increased the risk of suffering the pleaded cardiovascular conditions. In response to a request for particulars of his pleading, the applicant gave the following meaning to his allegation of a material increase in risk:
The increased risk is said to be material in that:
(i) the consumption of Vioxx elevated the chance of a person who consumed Vioxx experiencing one or more of the cardiovascular conditions; and
(ii) a reasonable person would be likely to attach significance to it.
The respondents took the applicant here to be invoking the test propounded in the joint judgment in Rogers v Whitaker (175 CLR at 490 − see para 784 above). They then went to that judgment, and noted that the hypothetical person referred to in subpara (ii) of the applicant’s response was not just any reasonable person but such a person in the patient’s position. From this it followed, according to the respondents, that the “ascertainment of the materiality of a risk involves an individual element”. Therefore, whether the respondents ought to have regarded a risk as material was not susceptible of a single answer across all group members, but would have to be determined on a case‑by‑case basis. It followed again from that that the only group member with respect to whom the court could now reach a conclusion as to the nature of the response reasonably required of the respondents to the circumstance (if it were a circumstance) that Vioxx increased the risk of suffering the pleaded cardiovascular conditions was the applicant himself.
799 I do not accept the respondents’ analysis of the law relating to the steps required of them in the discharge of their duty of care. Here one must start with the circumstances from which the group members’ cause of action arises. This is a case in which the product in question has been manufactured as intended, without the intrusion of a foreign substance, without any miscalculation in the mix of ingredients and without any deterioration in the course of wholesale or retail supply. The kind of considerations which, in such a case, should inform the inquiry were referred to by the UK Court of Appeal in Wright v Dunlop Rubber Co Ltd (1972) 13 KIR 255, 272, in a passage cited with apparent endorsement in the Appeal Division of the Supreme Court of Victoria in Thompson v Johnson and Johnson Pty Ltd [1991]2 VR 449, 490:
It is obvious that the answer to the question: ‘What are reasonable steps?’ must depend upon the particular facts. It is obvious, also, that the duty is not necessarily confined to the period before the product is first produced or put on the market. Thus, if, when a product is first marketed, there is no reason to suppose that it is carcinogenic, but thereafter information shows, or gives reason to suspect, that it may be carcinogenic, the manufacturer has failed in his duty if he has failed to do whatever may have been reasonable in the circumstances in keeping up to date with knowledge of such developments and acting with whatever promptness fairly reflects the nature of the information and the seriousness of the possible consequences.
If the manufacturer discovers that the product is unsafe, or has reason to believe that it may be unsafe, his duty may be to cease forthwith to manufacture or supply the product in its unsafe form. It may be that in some circumstances the duty would be fulfilled by less drastic action: by, for example, giving proper warning to persons to whom the product is supplied of the relevant facts, as known or suspected, giving rise to the actual or potential risk. Factors which would be relevant would be the gravity of the consequences if the risk should become a reality, and the gravity of the consequences which would arise from the withdrawal of the product.
800 The applicant does not allege that, as a direct and inevitable result, the consumption of Vioxx gave rise to any one or more of the pleaded cardiovascular conditions. Rather, his case is concerned with an activity by the respondents which is said to have given rise to a risk to him, and to the other group members, of which they knew or ought to have known. As reasonable manufacturers and suppliers of Vioxx, what ought the respondents have done in such circumstances? The answer, I consider, is to be found in the judgment of Mason J in Wyong Shire Council v Shirt (1980) 146 CLR 40, 47−48:
In deciding whether there has been a breach of the duty of care the tribunal of fact must first ask itself whether a reasonable man in the defendant’s position would have foreseen that his conduct involved a risk of injury to the plaintiff or to a class of persons including the plaintiff. If the answer be in the affirmative, it is then for the tribunal of fact to determine what a reasonable man would do by way of response to the risk. The perception of the reasonable man’s response calls for a consideration of the magnitude of the risk and the degree of the probability of its occurrence, along with the expense, difficulty and inconvenience of taking alleviating action and any other conflicting responsibilities which the defendant may have. It is only when these matters are balanced out that the tribunal of fact can confidently assert what is the standard of response to be ascribed to the reasonable man placed in the defendant’s position.
This was not a product liability case, of course, but the principles laid out by Mason J are, in my view, universal.
801 Conformably with the passage from Wyong v Shirt referred to above, the question in the present case is: what would the reasonable manufacturer and supplier do by way of response to the risk identified? I consider it to be a distraction to attempt to equate the position of the respondents with that of a surgeon advising a patient about the risks involved in undertaking a particular procedure. The respondents were not in a one‑on‑one relationship with the consumers of Vioxx. They were manufacturers and suppliers of a mass‑produced product, packaged and labelled by them without discrimination as between those consumers. Any decision they made about continuing to supply Vioxx, or about providing a warning, would have to be made generally (although, I accept, a warning may in its terms have made such a discrimination). They did not pretend to understand the circumstances of particular patients. Quite the contrary: by relying on the “learned intermediary” defence, they implicitly disclaimed any such understanding. The so‑called subjective limb of the two‑part test in Rogers v Whitaker can, therefore, have no relevance to the circumstances of the respondents in the present case.
802 In the particulars which he provided (as mentioned in para 798 above), the applicant confined himself to a modified version of the objective element of the test in Rogers v Whitaker. He contended that the risk in the present case should be regarded as material if a reasonable person would be likely to attach significance to it. I say “modified version” because, as will be noted, the applicant omitted the qualifier “in the patient’s position”. In so doing the applicant was proceeding consistently with the distinction I have made above between the circumstances of a surgeon advising an individual patient about the risks involved in a contemplated procedure and those of the respondents as manufacturers and suppliers of a product intended for general distribution. I consider that it would be quite artificial to impose upon the respondents an obligation to consider the individual situation of every consumer who did or might purchase Vioxx and to make individual decisions about whether to supply the drug to him or her, or whether to warn, and if so in what terms. Even an objective test, if posited on the “patient’s position”, would, in my view, be quite unworkable where those presumptively making an informed response to the risk as they reasonably perceived it were manufacturers and suppliers such as the respondents.
803 I consider that the matter should be approached in a much less complicated way than was proposed by the respondents. That approach should be as stated by Mason J in Wyong v Shirt: confronted with the existence, or the prospect, of a risk of which he or she knew or should have known, what would a reasonable person in the position of the respondents have done by way of response? The object of the reasonable person would be, of course, to eliminate or to minimize the scope for the risk coming home, or at least to warn the consumer, or to provide information or advice in the nature of a warning, so that he or she might weigh the size and nature of the risk, or the prospect of a risk, against the benefit of taking the drug, and make his or her own decision. If the reasonable person would have perceived that there were, or might be, differences as between consumers that would militate against a “one size fits all” approach, then no doubt an appropriate discrimination would be made in the terms in which any warning was expressed. However, as will appear, I do not accept that the nature of the response required of the respondents cannot be identified without assuming to them a knowledge of the individual circumstances of all consumers. Indeed, when a response to the results of the VIGOR trial was called for, MSDA took a single step upon which it now relies as constituting the discharge of its duty of care to all consumers (the amendment of the Product Information in November 2001). Likewise, the response of the respondents to the results of the APPROVe trial was a single one which applied across all patient groups (the withdrawal of Vioxx).
804 I turn now to the facts of the case, and to the question whether, in the circumstances obtaining, the respondents discharged their duty of care to consumers of Vioxx. I have held that there was no time before the withdrawal of Vioxx from the market when the respondents knew or ought to have known, as a matter of established scientific fact, that the consumption of rofecoxib materially increased the risk of suffering the pleaded cardiovascular conditions. However, I have also held that the results of the VIGOR trial ought to have been regarded by them as a worrisome signal of potential cardiovascular risk. As manufacturers with a duty of care, how ought the respondents reasonably have responded to that circumstance? I do not think – and the respondents did not submit – that doing nothing would have been a reasonable response.
805 What Merck ought to have done was the subject of an allegation in the Further Amended Statement of Claim, as follows:
At all material times after 30 June 1999, in manufacturing the rofecoxib from which Vioxx tablets were manufactured by Merck Australia, Merck Inc:
(a) failed to undertake or cause to be undertaken any or any adequate research, investigations, clinical trials or observational studies in order to ascertain the adverse side effects and health risks that may be associated with the consumption of rofecoxib including a material increase in the risk of suffering the [pleaded] cardiovascular conditions;
(b) failed to have adequate regard to the results of any research, investigations, clinical trials or observational studies undertaken or caused to be undertaken by Merck Inc or others suggesting that the consumption of rofecoxib materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(c) failed to await the results of any research, investigations, clinical trials or observational studies being undertaken or planned by Merck Inc or others to ascertain whether the consumption of rofecoxib materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(d) withheld from and failed to publish in medical publications or otherwise reveal data and information of which it was aware concerning adverse cardiovascular risks associated with the consumption of rofecoxib.
It follows from my reasons above that I reject the allegations set out in subparas (a) and (b) of this pleading.
806 The allegation in subpara (d) was the subject of little elaboration in the applicant’s final submissions, or in his case generally. No attempt was made to establish the proposition that the discharge of the duty of care which lay upon Merck involved a positive obligation to “publish” data and information. However that may be, on no view (subject only to what I mention in the next paragraph) did Merck fail “otherwise to reveal” all the data and information about the cardiovascular results of the various trials and studies of Vioxx with which it was associated. There were no relevant data to which the applicant could point in the present case which had not been provided to the FDA and TGA. In the way he conducted his case, the applicant put in issue not the failure to publish the data and information, but Merck’s interpretation of the data and information which was available to it.
807 The one instance where Merck’s omission to publish something was controversial related to the three additional myocardial infarctions which were not included in Bombardier (2000) as submitted to the New England Journal of Medicine (see paras 138‑142 above). While the applicant was critical of that, and used to some forensic effect the expression of concern by the editors of the Journal, ultimately it is hard to see how these circumstances bear upon the question whether Merck (or, for that matter, MSDA) discharged its duty of care. As pointed out in the correspondence which followed the expression of concern, whether with or without the three myocardial infarctions, the data revealed that there was a statistically significant difference between the risk of taking Vioxx and the risk of taking naproxen. Further, however disputable as a matter of publishing ethics, the authors of Bombardier (2000) had a credible explanation for the non-inclusion of these events that I am in no position to hold to be unreasonable (ie that they were reported after the cut-off date). Neither was it as though Merck made any secret of the full extent of the relevant data: they were included in the safety update report to the FDA of 13 October 2000.
808 I would also note that the applicant’s allegation in relevant respects is that Merck’s dereliction was in substance one of commission, notwithstanding his use of the familiar formula “failed to …”. I say this because there could be no suggestion that Merck would have fallen short in the discharge of its duty of care to group members in this proceeding had Bombardier (2000) not been published at all. The vice, to the extent that there was one, is that a misleading impression of the extent of cardiovascular risk was conveyed. Although I am in a sense anticipating an aspect of the applicant’s negligence case to which I shall turn below (causation), I consider it unlikely in the extreme that any difference would have been made to the clinical judgments made by the cohort of medical practitioners whose decisions are presently relevant if the three additional myocardial infarctions had been used as a basis for the rates set out in the article (particularly given the very small impact which that would have made thereby, as pointed out by Drs Reicin and Shapiro − see para 141 above).
809 The kernel of the applicant’s pleading is, of course, the allegation in subpara (c) in the extract from the Further Amended Statement of Claim set out in para 805 above. Expressed in a positive way, here the applicant is alleging that Merck ought not to have made rofecoxib available for incorporation into Vioxx until the completion of any relevant research, investigations, clinical trials or observational studies. This aspect was stressed by counsel for the applicant in their closing submissions. While recognising that, until September 2004, it had not been established that Vioxx caused or contributed to CVT events, they submitted that Merck ought nonetheless to have recognised that there was a risk that Vioxx was working in that way, and that such a cause or contribution would ultimately be scientifically established. They submitted that, in the meantime, Merck ought not to have continued to supply rofecoxib to MSDA for incorporation into Vioxx tablets.
810 I accept so much of the applicant’s submission as involves the proposition that Merck ought to have recognised that it might ultimately be found that the consumption of Vioxx caused or contributed to CVT events. In the state of the science as it existed until September 2004, however, I am not persuaded that the only course reasonably open to the respondents was to have withdrawn Vioxx from the market altogether; or that the only course reasonably open to Merck was to have made rofecoxib unavailable to MSDA. Rofecoxib was not a drug which, as a direct and inevitable consequence, caused CVT events in all cases. Indeed, the occurrence of CVT events in the Vioxx arm of the VIGOR trial (including those which presumably were not caused or contributed to by Vioxx) was only 1.67 such events for every 100 patient‑years. As against this, I must recognise that Vioxx was a drug which worked a lot of good: for many people (including the applicant) it brought relief from the pain and discomfort associated with arthritis without gastrointestinal side‑effects. As a prescription drug, it required the judgment of a medical practitioner before any person could consume it. For rofecoxib to have been withdrawn upon nothing more than the signal of potential risk generated by VIGOR would have been to deny a great many arthritis sufferers a means of securing comfort which they may freely have chosen, even if fully informed. It is notorious that many drugs carry side‑effects, yet resort is had to them as the result of the balancing by patients of the risks and benefits to be derived, informed and assisted by the professional judgments of their medical practitioners.
811 In the circumstances as they existed until September 2004, the present was a case in which Merck’s duty of care as manufacturer and supplier of rofecoxib could be discharged by the provision to its subsidiaries – in the present case, MSDA – of information necessary to enable the latter to formulate and to disseminate adequate instructions and warnings about the possibility of the drug causing or contributing to CVT events. In the present case, MSDA did not suggest that it was unable, by reason of lack of information from Merck, to provide the appropriate instructions and warnings. Indeed, the respondents’ case was based upon the silent proposition that MSDA should be fixed with the whole of Merck’s relevant scientific knowledge at each point in time. On all of the evidence in the case, I am satisfied that that was a realistic position to take, and one which reflected the facts as they existed. That being the case, I think that Merck had done everything that might reasonably be expected of it in the discharge of its duty of care.
812 The applicant did allege that Merck failed to provide to pharmacists, to medical practitioners and to other health care professionals, and to the Australian public, any or any adequate information, advice or warning to the effect that the consumption of rofecoxib materially increased the risk of suffering the pleaded cardiovascular conditions. However, the obligation to provide information, advice or warning of the kind referred to fell upon MSDA as the manufacturer and supplier of Vioxx in Australia. Merck would, in my view, have discharged its duty of care by ensuring that MSDA was fully informed about the nature and biological operation of rofecoxib, and about the existing state of the science with respect to the consequences, in the vasculature, of consuming it. I am satisfied that it did so.
813 For the above reasons, I propose to dismiss the applicant’s case in negligence against Merck.
814 The applicant’s negligence case against MSDA commences with allegations that MSDA −
(a) utilised a non‑steroidal anti‑inflammatory drug, namely rofecoxib, the consumption of which materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(b) failed to utilise a non‑steroidal anti‑inflammatory drug the consumption of which did not materially increase the risk of suffering the [pleaded] cardiovascular conditions;
(c) failed to make any or any adequate inquiries of Merck … regarding the adverse side effects and health risks that may be associated with the consumption of rofecoxib including a material increase in the risk of suffering the [pleaded] cardiovascular conditions.
The applicant then makes substantially the same allegations against MSDA as those set out in subparas (a), (b) and (c) of his allegations against Merck as set out in para 805 above. With respect to those allegations, I find, for the same reasons as I have given above in relation to Merck, that MSDA did not breach its duty of care merely by using rofecoxib in Vioxx rather than some other NSAID. As to (c) in the extract above, very little attention was paid to this aspect in the applicant’s case. As I have said, I infer that MSDA was as well‑informed about the potential side‑effects and health risks of Vioxx as was Merck itself. The resources and research of MRL were available to MSDA, whose management were in regular contact with Merck. I do not think there is anything in the applicant’s “failure to inquire” case.
Failure to warn
815 The next part − and a very significant part − of the applicant’s case is centred on his “failure to warn” allegations. He alleges that MSDA failed to provide any or any adequate information, advice or warning to the effect that the consumption of rofecoxib or of Vioxx tablets materially increased the risk of suffering the pleaded cardiovascular conditions. He so alleges in a number of contexts. He says that no such information etc was set out on the packets in which Vioxx was sold, or on the labelling associated therewith. He says that no such information etc was provided to pharmacists, medical practitioners and other health care professionals, either in the marketing, or in the distribution and supply, of Vioxx. And he says that no such information etc was provided to the general public (being the consumers or potential consumers of Vioxx tablets) or to pharmacists, medical practitioners and other health care professionals who might have advised or treated members of the public. He also alleges that Merck failed to provide such information etc to MSDA, to pharmacists, medical practitioners and other health care professionals, or to the public. All these allegations are denied.
816 The applicant’s “failure to warn” case implicitly calls up the following questions: first, did a duty to warn arise, and if so when, and by reference to what circumstances; secondly, of what risk ought the warning to have been given, to whom and in what terms; and thirdly, did MSDA provide a warning which, in its timing, content and communication ought to be regarded as having discharged the duty of care which was owed to consumers of Vioxx?
817 As to the first question, it was not seriously suggested on behalf of the applicant that a duty to warn arose until the preliminary results of the VIGOR trial were available in March 2000. Other aspects of the alleged tendency of Vioxx to be associated with cardiovascular disease did not feature in the applicant’s failure to warn case. It was not suggested, for example, that MSDA was derelict in not having issued a warning that Vioxx was associated with hypertension. Had it been, it is difficult to see how such a case might have been sustained, given the terms of the original Product Information (see para 225 above) that was sent to doctors by correspondence in October 2000 (see para 226 above). These possibilities, however, do not have to be considered further, as they were not elements of the applicant’s case. To the extent that it is necessary to do so explicitly, I hold that no duty to warn arose before the arrival of the preliminary VIGOR results in March 2000.
818 Did the results of the VIGOR trial give rise to a duty to warn? There are, in my view, two reasons why it should be held that they did. First, as noted elsewhere in these reasons, those results represented a worrisome and important signal of potential cardiovascular risk, and ought reasonably to have been so viewed by the respondents. As I have held, Merck did so view them. The respondents’ case was conducted without discrimination as between Merck and MSDA on matters of scientific knowledge and understanding. They called no member of the medical or scientific staff of MSDA, in which event I would be prepared to infer that MSDA viewed the VIGOR results in the same way as did Merck. However that may be, I would hold that MSDA ought, viewing the matter reasonably and prudently (as is appropriate in matters of safety), to have taken the same view as expressed by the cardiologists, namely, that the results were a worrisome and important signal of potential cardiovascular risk.
819 That view reasonably mandated, in my opinion, the giving of a warning. It was, as I have held, a reasonable course for Merck to have undertaken a cardiovascular outcomes study, and MSDA was entitled to rely on the ongoing work of MRL in that regard. Neither was it necessary that Vioxx be withdrawn from the matter pending the results of such a study. However, in the meantime it was, I consider, incumbent on MSDA (ie consistently with the discharge of its duty of care) to issue a warning − or some information or advice in the nature of a warning − which reflected the state of understanding which arose from the VIGOR results.
820 Secondly, it was ultimately accepted by MSDA that a warning in appropriate terms was necessary to be introduced into the “Precautions” section of the Vioxx Product Information. It is true that the revised Product Information that was originally submitted by MSDA in January 2001 did not contain any such warning, but the clinical evaluator was – it seems quite emphatically – of the view that one was necessary in the circumstances. The then delegate agreed. So do I. Ultimately, MSDA itself accepted that there had to be a warning in the Product Information. In the light of this history of events, it could hardly be suggested that no warning at all was called for in consequence of the results of the VIGOR trial.
821 As I have set out at paras 721‑723 above, Dr Dickman was cross‑examined about his knowledge of the general subject of Vioxx and cardiovascular risk as derived from sources other than MSDA. I do not understand the respondents presently to be proposing, however, that such sources as I there referred to should be regarded as satisfactory substitutes for an appropriate warning from MSDA itself. I would certainly not so regard them. They may be relevant to the matter of causation, but they do not, in my view, bear upon the question whether MSDA took such steps as would be sufficient to discharge its duty of care.
822 When should a warning have been given? When the VIGOR preliminary results became available in March 2000, Vioxx had secured registration on the ARTG, but was not yet formally on the market in Australia. Vioxx was announced to the medical profession, and a copy of the Product Information provided, by way of Ms Paterson’s letter of October 2000 (see para 226 above). By then, the VIGOR Clinical Study Report had been forwarded to the FDA (on 29 June 2000). Indeed, what were effectively the final cardiovascular results of VIGOR were forwarded to the FDA in the safety update report of 13 October 2000. MSDA was, therefore, possessed of sufficient information to provide the necessary warning no later than its letter of October 2000 formally announcing the availability of Vioxx. In my opinion it should have done so.
823 Turning to the second question identified in para 816 above, quite obviously it was with cardiovascular risk that a warning should have been concerned. However, as the respondents pointed out, at no stage did the applicant formulate the actual terms of a warning that should have been given. Rather, he contended that “MSDA … [was] negligent in failing to provide any adequate information, advice or warning to the effect that the consumption of Vioxx materially increased the risk of suffering the [pleaded] cardiovascular conditions”. I am prepared to work on the basis that the kind of warning which the applicant proposed should have been incorporated into the Product Information in 2000 would have been along these lines.
824 The imposition on MSDA of a duty to warn in such terms would be problematic in a number of respects. First, the “signal” given by VIGOR was not a consistent one over all of the pleaded cardiovascular conditions. To say that the consumption of Vioxx might have increased the risk of suffering each of those conditions would have been to overreach the results of VIGOR on any view. Secondly, the VIGOR results did not speak at all along the Vioxx‑placebo axis. The only sense in which they justified a statement about increased risk was with reference to a situation in which the notional comparator was naproxen. Thirdly, the trial was not concerned with, and (as the cardiologists in the present proceeding agreed) said nothing about, a situation in which Vioxx was taken at the recommended maximum therapeutic dose in Australia − 25 mg daily. Fourthly, a warning in such terms as proposed by the applicant would have gone beyond what, as would reasonably have appeared to the respondents, was the prudent scientific consensus in 2000/2001. Here I refer to the views of the FDA Arthritis Advisory Committee expressed on 8 February 2001 − see para 156 above. And fifthly − and perhaps most importantly for present purposes − a warning in the terms proposed by the applicant would have gone beyond what I have held the respondents either knew or ought to have known about the consequences of consuming Vioxx until September 2004. I take the view, therefore, that the availability of the VIGOR results did not oblige MSDA to issue a warranty in the categorical terms proposed by the applicant.
825 Does that conclusion mean that there was no warning that MSDA could have issued, and acting reasonably should have issued, that would have fairly reflected the results of the VIGOR trial? I do not think so. Here it is convenient to commence with the warning which did ultimately find its way into the “Precautions” section of the Vioxx Product Information in November 2001 (see para 258 above). Would that have been a sufficient warning if given in October 2000? The applicant submitted that the relevant paragraph in the Product Information was inadequate as a warning. To provide an opinion about that paragraph (and other things), he led the following evidence from Prof Donovan:
In my opinion, the language used in this product information does not convey a warning that Vioxx causes, or could cause, an increased risk of cardiovascular thrombotic events. The framing of the results is consistent with the reduction framing method described above and employed elsewhere in Merck marketing messages. Although the “naproxen theory” is not explicitly propounded, the reference to the difference in antiplatelet activity between some COX-I inhibitors and COX-2 selective inhibitors provides an explanation for the VIGOR cardiovascular events that exonerates Vioxx as a cause of increased CVT events. … The explicit statement that “In other controlled clinical trials spontaneous reports of these cardiovascular events are similar between Vioxx and nonselective NSAID comparators” precludes any interpretation of an increased risk with Vioxx. The statement then appears to minimize the clinical significance of any adverse findings by simply providing generic advice to doctors to “assess the importance of these data” when prescribing for patients at CV risk.
826 Prof Donovan was cross‑examined on this evidence. He accepted that he had not undertaken any specific study on the subject of how a prescribing practitioner would interpret the paragraph. He agreed that whether or not the paragraph conveyed a warning would depend partly upon the knowledge and expertise of the person reading it. When it was put to him that the statement that “the difference in antiplatelet activity between some COX-l inhibiting NSAIDs and COX-2 selective inhibitors may be of clinical significance in patients at risk of thrombo‑embolic events” stood as a clear warning that the difference in antiplatelet activity as between Vioxx and some NSAIDs might have a consequence in the particular clinical situation with which a doctor was confronted, he said: “I don’t know that it’s a clear warning but it’s certainly a clear statement of the data.” When it was suggested that the words stood as a clear signal to doctors to proceed with caution, Prof Donovan said:
Well, I don’t know how doctors would interpret this. I have an opinion of whether or not it sends a message of increased cardiovascular risk but I can’t say what an individual doctor – how they would interpret that.
827 Dr James Lynch was a very experienced general practitioner called by the applicant as an expert. In his witness statement, he said that he had been asked to provide an opinion, based on his training, knowledge and experience in general practice, about the Product Information that accompanied Vioxx. He referred to the Product Information that was approved by the TGA in November 2001, and to the passage in it to the effect that Vioxx was not a substitute for aspirin for cardiovascular prophylaxis because of its lack of platelet effects. He interpreted this to mean that, if patients were taking aspirin, they should continue to do so after commencing Vioxx, not as a warning that Vioxx might have given rise to a cardiovascular risk. He referred to part of the section headed “Cardiovascular Effects”, but made no (admissible) comment about it.
828 Dr Dickman did not express an opinion about the adequacy of the warning in that section of the Vioxx Product Information. Neither did Dr Anderson.
829 In his witness statement, Dr Bertouch said that the paragraph contained “an appropriate cautionary note regarding the difference in cardiovascular thrombo‑embolic events between VIOXX and naproxen”. Save by way of drawing attention to the circumstance that patients do not normally view the Product Information, Dr Bertouch was not challenged on this evidence.
830 As a communication addressed to a professional audience, I consider that the new paragraph inserted into the Vioxx Product Information in November 2001 was a reasonable warning of the potential cardiovascular risk disclosed by the VIGOR results. It drew attention to difference in CVT events as between the two arms of the study, and to the fact that the comparator was naproxen. It accurately identified the cohort of patients that had been involved in the trial, and the dose that had been used. It referred to other clinical data where no difference had been disclosed as between Vioxx arms and (non‑naproxen) NSAID arms. And it drew attention to the critical biochemical difference between coxibs and non‑selective NSAIDs, namely, the absence of any antiplatelet activity in the case of the former. As to this last aspect, it is important to note that the clinical context in which Vioxx might be under consideration was one in which the patient was presumptively suffering from osteoarthritis and would, normally, be in need of an anti‑inflammatory drug. The paragraph in the Product Information was such as would draw the prescriber’s attention to the difference between coxibs and at least some other NSAIDs, most obviously naproxen, with respect to antiplatelet activity. Whereas all NSAIDs inhibit COX-2 (and, as Prof Celemajer said, are given in doses that achieve that purpose wholly or very substantially), traditional NSAIDs also inhibit COX-1, thereby suppressing, to an extent at least, the aggregation of platelets. Vioxx did not. This was the essence of the distinction between Vioxx and the traditional NSAIDs. It was the distinction which would have been relevant to the clinician; and it was the distinction to which the new paragraph in the Product Information pointed.
831 The other consideration is that the Product Information was a medical document intended to be read by medical practitioners. It is, therefore, substantially by reference to the evidence of the medical witnesses that the court should attempt to assess the sufficiency of the Product Information as a warning of such risks as ought reasonably to have been apparent to MSDA in the period subsequent to VIGOR. As will be apparent from what I have said above, no medical witness expressed the view that the terms of the paragraph inserted in November 2001 were less than sufficient for that purpose. Dr Bertouch considered that the paragraph sounded “an appropriate cautionary note”. In these circumstances, unless it was, as a matter of ordinary language, palpably inadequate as a warning, I would be most reluctant to hold that the paragraph, in the reading of it through professional eyes, did not fairly raise the prospect that the consumption of Vioxx might be associated with the onset of CVT events. For reasons stated earlier, I do not think it was palpably inadequate.
832 Finally with respect to the reasonableness of the paragraph in the Product Information that was introduced in November 2001, I note that that paragraph was approved by the TGA. It is true that its wording owed much to the input of MSDA, and I accept the applicant’s point that MSDA made every effort to render inconspicuous the message that Vioxx might well be pro‑thrombotic. However, the differences between the paragraph which came to be included in the Product Information, and that recommended by the clinical evaluator on 3 July 2001 (save with respect to the co‑administration of aspirin) are, to my reading of them, essentially matters of emphasis and balance. The original delegate, Dr Anantharajah, recommended that the Product Information should contain a statement that “long‑term therapy is not recommended for those with a history cardiovascular disease”, which would not, in my estimation, have been much different in point of substance from that ultimately adopted: “Physicians should assess the importance of these data for an individual patient at risk for cardiovascular thrombo‑embolic events, if considering long‑term therapy with a COX-2 selective inhibitor”. I make this comparison because the applicant was not critical of the evaluator or of Dr Anantharajah.
833 To whom should such a warning have been communicated? If, as I have held above, a warning in the terms of the new paragraph inserted into the Product Information in November 2001 should be regarded as sufficient, clearly such a warning should have been communicated at least to general and other medical practitioners who might reasonably be supposed to have had cause to treat or to advise patients suffering from osteoarthritis (and, subsequently to 17 January 2002, from rheumatoid arthritis). Had this been done, I do not consider that a warning to pharmacists would have been required in addition. The nature of such a warning would have implied the making of the necessary professional judgments by the prescribing practitioner. Once a decision to prescribe Vioxx had been made, I do not think that MSDA ought reasonably to have contemplated a further monitoring role for the dispensing pharmacist. MSDA should not be regarded as having fallen short in the discharge of its duty of care by not putting the pharmacist in possession of a warning in the same or similar terms as those provided to the prescriber.
834 The applicant’s case as pleaded proposed that the warning ought also to have been communicated to “other healthcare professionals”. This proposal was, however, developed neither in the evidence nor in the applicant’s final submissions. I do not know who these other professionals might have been. I do not think that the applicant has made good his case in this regard.
835 The applicant also alleged that MSDA ought to have placed a warning (or “information” or “advice” as to cardiovascular risk) on the Vioxx packets and labels. One of the oddities of this case is that, despite the mountain of evidence that was tendered, neither a Vioxx packet nor any example of the labelling used on Vioxx was placed into evidence. The applicant submitted that the packet and label contained no warning, but the only evidence as to that aspect was the applicant’s, that he read the label or product information when he was first put on Vioxx, but did not recall reading anything that suggested or implied that Vioxx would increase his risk of suffering a heart attack. He was not challenged on this evidence (although, as noted elsewhere, he clarified that the “product information” to which he referred was the package insert, or “Consumer Medicine Information”). I regard it as having been established in the applicant’s own case, therefore (but not otherwise), that MSDA did not include any presently relevant warning on the packets in which Vioxx was sold, on the labelling associated therewith or as part of the Consumer Medicine Information.
836 If an appropriate warning had been provided to medical practitioners, however, I do not think that MSDA fell under any duty additionally to warn the patient, or consumer. Once a professional judgment had been made to prescribe Vioxx, with all of the balancing and compromises that that implied, I consider that the addition of a warning to which the patient would first be exposed when he or she obtained Vioxx from the pharmacist would be both inappropriate and unnecessary − not to mention somewhat confusing. The contradictions involved in allowing for Vioxx to be prescribed by a doctor and then recommending that the patient himself or herself make a further judgment based on his or her own assessment of underlying cardiovascular risk were not addressed by the applicant. Had he been serious about his submission that MSDA fell short in the discharge of its duty of care by not providing such a warning, they should have been.
837 To date in this section of my reasons, I have held that MSDA ought to have warned medical practitioners of the signal yielded by the VIGOR results at the time when Vioxx was introduced on to the Australian market in October 2000. A warning in the terms set out in the new paragraph in the “Precautions” section of the Product Information in November 2001 would have been sufficient. I do not hold that, to be effective, a warning had to be in those terms. I mention them because they represent a formulation with which MSDA was content and which would have gone no further than the state of the science justified. Was such a warning, or any equivalent warning, ever given?
838 On no view was any warning given to medical practitioners by MSDA before November 2001. In its final submissions, MSDA did not directly confront this omission. Indeed, it is implicit in its submissions that there was no such warning. However, it is also implicit that the Product Information was a sufficient warning to doctors of the signal of cardiovascular risk presented by the VIGOR trial. This is a question which arises only upon consideration of the situation as at November 2001, and I shall return to it. Before then, there was nothing in the Product Information which would have put doctors on notice of that signal. MSDA justified the absence of any reference to the VIGOR results by the need to obtain the approval of the TGA for the terms of an amendment to the Product Information. The applicant submitted, however, that a safety‑related amendment could have been made at any time. This submission was well‑founded (see paras 205 and 207 above), but the respondents rejoined that the amendment which was in fact (finally) approved by the TGA would not have been permissible as a safety‑related one because it was based upon, and involved an explanation of the results of, a clinical trial which compared Vioxx with another therapeutic good (naproxen). The respondents submitted that this would not have come within the terms of s 9D(2) of the TG Act. In this they were mistaken, since s 9D was not part of the TG Act until October 2002. However, the provision that became s 9D(2) was s 32(4) of the TG Act before then (see para 205 above).
839 As pointed out elsewhere in these reasons, the effect of s 32(4) was to require the Secretary to vary the terms of a Product Information where (amongst other things) the only effect of the variation would have been to add a warning or precaution that did not include a comparison with other therapeutic goods. But the provision did not prohibit the Secretary from allowing a variation even where such a comparison was involved. According to Mr Back, when he first informed Dr Chipman of the results of VIGOR in March 2000, the latter said that MSDA should immediately consider a safety‑related notification, and that the TGA would be happy to include important safety information in the current Product Information, no matter what the patient group. MSDA did not so proceed, for which omission there was not, I consider, a satisfactory explanation. However, since the making of an approved amendment to the Product Information which reported on the signal of risk yielded by the VIGOR results in a way which did not overreach, and which did not do unnecessary injustice to Vioxx, would have required the approval of the TGA, an organisation which MSDA did not control, I am not prepared to hold that MSDA might, by confining its amendment to matters of safety, have procured such an amendment very promptly in mid‑2000 or thereabouts.
840 However, the Product Information was not the only means by which MSDA might have communicated with medical practitioners. The evidence justifies the conclusion that it often communicated by way of “Dear Doctor” letters which, I infer, could readily have been sent to all relevant practitioners at any time. Indeed, it was such a letter that formally announced the availability of Vioxx in October 2000. That letter − which is reproduced in para 226 above − contained a warning about possible interactions with warfarin, methotextrate and lithium. If there is any reason why that letter − or another letter sent out at about the same time − might not have conveyed an appropriate warning about the signal of cardiovascular risk yielded by VIGOR, none was suggested either in the evidence or in the respondents’ submissions. I do not, therefore, accept that MSDA’s inability to procure an amendment to the Vioxx Product Information before November 2001 is a sufficient explanation for its failure until then to provide medical practitioners with an appropriate notification of the VIGOR cardiovascular risk signal.
841 When the Vioxx Product Information was eventually amended to reflect the results of the VIGOR trial − in November 2001 − did that constitute a sufficient warning of the cardiovascular risk signal? I have held that it was sufficient in its terms. But was the manner of its communication such that MSDA was reasonably entitled to assume that all medical practitioners would see it and take note of it? This is a difficult and finely‑balanced question. There were certain propositions which, expressly or implicitly, informed the respondents’ case on negligence: that the Product Information was the, or at least the most important, means by which MSDA communicated with the medical profession; that the Product Information was controlled by the TGA (in the sense that, as the respondents put it, the TGA was the “final arbiter” of the terms thereof); that (for that reason) it could not be said that MSDA itself was free to communicate with doctors (or others) in terms of its own choosing; and that knowledge of the Product Information by doctors (as amended from time to time) should be presumed, much as one would presume knowledge of the terms of statutes, for instance. Upon analysis, however, these propositions are at best problematic and in some respects unfounded.
842 As I have mentioned elsewhere in these reasons, the TG Act did not give the Product Information any such status as the respondents’ submissions implied. Indeed, it did not treat product information as, or as the subject of, a document which would have a discrete existence. The term “product information” was used once only (in s 32) as a shorthand reference to “information relating to the safe and effective use of the goods, including information regarding the usefulness and limitations of the goods” included in the entry in the ARTG that related to the goods in question. Even then, the term was used only in a provision which identified the circumstances in which the Secretary was obliged to vary certain details in the ARTG. It was the 1994 guidelines, approved for the purpose of s 23(2), which gave the “Product Information” its identity as a discrete document and which required it to contain the full range of details set out in para 208 above. That requirement of the TGA, together with conditions established under s 28(2), gave the Product Information its important place in the regulatory scheme for therapeutic goods in Australia.
843 I was not referred to any provision − and for my own part I have found none − which required the Product Information to be published or disseminated, either to medical practitioners or at all. It seems, however, that the Product Information for any drug on the ARTG was accessible by doctors, at least by way of MIMS and such software as “Medical Director”. Although I was not told about it, I infer that that was a computer program available to doctors by private acquisition, much as, for example, they might obtain accounting software for their practices. Merely by providing Product Information to the TGA (and by amending it as appropriate) and by having it approved, however, a manufacturer in the position of MSDA did not either communicate with doctors or initiate a process of publication by which such a communication would occur in the inevitable course.
844 However, was the Product Information so firmly established as a resource readily available to medical practitioners that MSDA, acting reasonably and prudently in the discharge of its duty of care, was entitled to assume that, by doing no more than obtaining the TGA’s approval to an amendment thereof, it effectively communicated the terms of such an amendment to all such practitioners? It is clear that the Product Information, as it existed from time to time, was in fact readily available to all such practitioners. It may be that MSDA was entitled to assume that practitioners who contemplated the prescription of a new drug for the first time would, as a matter of course, look up the Product Information. However, Vioxx had been on the market in Australia for nearly a year in November 2001, and practitioners had presumptively read Ms Paterson’s letter of December 2000, in which some precautionary notes were sounded, but nothing was said about cardiovascular effects. Was such an assumption warranted in the circumstances of November 2001?
845 In his witness statement, Dr Dickman said that he would not be aware of a change to a drug’s Product Information unless notified about the change. He was cross‑examined about the updating of Product Information, but not specifically about this evidence. I should set out in terms what I perceive to be the relevant aspects of that cross‑examination:
So far as you know, is it the position that Medical Director is updated via some electronic means four times a year?---Yes.
And such new information as is necessary to be fitted into the program is sent down about four times a year?---Yes.
So far as the drugs are concerned, does it give you a note of what drugs have changed?---No, it doesn’t.
Give you a note if an indication has changed?---No, I don’t think so.
Prior to having MIMS over the Medical Director software, MIMS would be updated in paper three or four times a year, wouldn’t it?---Yes.
The updating version of MIMS would only contain those drugs that needed to be updated. Is that right?---Yes.
So you would know three or four times a year from your paper copy of MIMS what
drugs were being updated, is that right?---Yes.
Do you still keep a paper copy of MIMS in your surgery?---Yes.
Is that updated four times a year still?---Some of the copies are slower to get to us but I think, yes.
But there is a regular updating process which by looking at the index you can see what the drugs are that have been updated, is that right?---Yes.
That’s one way in which you would be notified about a change, isn’t it?---Yes.
Another way in which you would be notified about a change would be if a drug representative told you that the drug was now approved for a different indication?---Yes.
So, as an example, with Vioxx, it was initially approved for osteoarthritis, it was later approved as an indication for rheumatoid arthritis; that would be made known to you?---Yes.
When you came next to prescribe the drug, particularly if it was for someone with rheumatoid arthritis, you would go back to the product information and look to see with respect to that indication what the relevant information was, wouldn’t you?---Yes.
Of course, you may hear about a change from one of your colleagues. Is that right?---Yes.
Or you may get it from one of your other sources of information; a specialist noting that something has changed by changing a prescription, is that right?---Yes.
Or through one of the other sources that you have spoken about. Is that right?---That’s right.
846 I do not think that Dr Dickman’s evidence travels the distance necessary to justify the conclusion that MSDA was entitled to assume that, as a matter of inevitable course, general practitioners would read the Vioxx Product Information amendment of November 2001. First, he was not challenged directly on the evidence in his witness statement to which I have referred. Secondly, the furthest he was pressed in cross‑examination was to accept that resources were readily available to him from which he could ascertain which drugs had recently had their Product Information updated: he was not induced to give evidence to the effect that he did keep abreast of such changes as a matter of course. And thirdly, it may be one thing to know which drug’s Product Information had recently changed: it would be quite another to be alerted to those changes that involved new precautions on potentially serious side‑effects.
847 Dr Lynch referred to, and was cross‑examined about, the Product Information (both generally and for Vioxx), but not in any way that was presently useful. Dr Anderson, a colleague of Dr Dickman’s at Tower Hill who was called by the applicant because one of the applicant’s prescriptions for Vioxx was written by him, also gave no presently useful evidence.
848 It follows that Dr Dickman’s was the only evidence that usefully dealt with the question of how a general practitioner would respond, as a matter of normal practice, to changes in the Product Information for a particular drug. As appears from what I have written above, I do not think it has been established that MSDA was reasonably entitled to assume that a practitioner would, without specific prompting, necessarily have taken note of the fact that the Vioxx Product Information had been amended, or of the content of the amendment. It was never suggested to Dr Dickman that he had been remiss in not having done so, or was thereby acting inconsistently with the accepted norms of his profession. In the result, his evidence in chief that he would not be aware of a change to a drug’s Product Information unless notified about the change stands unchallenged, and is not inconsistent with any oral evidence which he gave.
849 Clearly the making of an amendment to the Product Information should be regarded as a necessary response by a drug manufacturer to the results of a clinical trial which conveyed a signal of risk. However, I do not consider that such an amendment should of itself be regarded as a sufficient response − that is to say, as a sufficient discharge of the manufacturer’s duty of care. Absent the manufacturer taking steps directly to inform medical practitioners of the amendments, I do not accept that the manufacturer would be entitled to assume that every such practitioner − including the one directly involved in prescribing the drug for the putative applicant − would inevitably become aware thereof. Thus I would hold that, by doing no more than amending the Vioxx Product Information in November 2001, MSDA had not taken reasonable steps to warn medical practitioners of the cardiovascular risk signal thrown up by the VIGOR results.
850 Did MSDA in fact do more than that? It led no evidence of a direct communication to doctors on the subject. I have found that the pro forma “Dear Doctor” letter dated December 2001 was not sent (see para 267 above). I make these findings generally (ie not merely in the applicant’s case). Whether, apart from them, a particular doctor was warned (advised, informed etc) is a question upon which it will be open to the respondents to lead evidence in that part of the proceeding which relates to his or her patient. In the remaining sections of this part of my reasons, I deal with the applicant’s own circumstances.
851 Did Dr Dickman in fact come to read, or otherwise to learn of, the new paragraph in the “Precautions” section of the Vioxx Product Information of November 2001? I would find not. Against his evidence in chief that he would not, unless notified, be aware of a change to a drug’s Product Information, he was cross‑examined, if I may so observe with respect, rather tangentially to the important question whether he read the new paragraph in the Product Information. He was not asked that question directly. Counsel appeared to be more concerned to discover whether Dr Dickman knew “the ideas encapsulated” by the paragraph (which he did, at some time between 2001 and 2004 − see para 726 above). However that may be, I could not discern in any evidence given by Dr Dickman that, in November 2001 or thereabouts, the amendment to the Product Information came to his attention.
852 I would hold, therefore, that the applicant has made good his case that MSDA failed to warn his treating practitioner of the cardiovascular risk signal from VIGOR of which it had been aware since March 2000.
853 For the sake of completeness, I would add that it was part of the applicant’s pleaded case that MSDA failed to provide, to the general public, any (or any adequate) information, advice or warning to the effect that the consumption of Vioxx materially increased the risk of suffering the pleaded cardiovascular conditions. That allegation was denied by the respondents. A submission which went little further than the allegation in his pleading was made by the applicant in his final written submissions. The respondents made no response to that submission. Neither did they deal with the allegation in their own final written submissions. The matter was dealt with barely, tangentially and indirectly in the applicant’s case. Save to refer to a media release of 16 November 2001 announcing the publication, and generally the findings, of Konstam (2001), the applicant referred me to no evidence by reference to which I might uphold this allegation. It was as though, having made the allegation of a failure to act, it then lay upon the respondents to prove the contrary. Clearly it was not. The allegation was part of the applicant’s case, and, save in the context of his own personal claim (as to which see para 729 above), it has not been made good. That is not to say that I could find that MSDA did provide any such warning to the general public as the applicant alleged it failed to do: there is just no evidence about it one way or the other.
MSDA’s marketing representations
854 The applicant alleges that MSDA developed and implemented a marketing strategy or campaign, by which it formulated and disseminated to pharmacists, medical practitioners and other health care professionals the following “information about Vioxx”:
(a) Vioxx has an excellent gastrointestinal safety profile compared with other … NSAIDs …;
(b) Vioxx has an excellent overall safety profile compared with other NSAIDs;
(c) Vioxx does not increase the incidence of adverse cardiovascular events;
(d) higher rates of myocardial infarction and other cardiovascular events among persons taking Vioxx relative to persons taking other traditional NSAIDs (in particular naproxen) did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events but were attributable to the fact that the consumption of naproxen produced a lower incidence of cardiovascular events because naproxen inhibits platelet aggregation in a manner similar to aspirin while Vioxx does not;
(e) the higher rates of myocardial infarction among persons taking Vioxx relative to persons taking naproxen observed in the Vioxx Gastrointestinal Outcomes Research trial … did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events;
(f) any information to the effect that Vioxx was not as safe as other NSAIDs because the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleasded] cardiovascular conditions was incorrect, misleading, exaggerated and/or unreliable.
The applicant describes these things as “the Vioxx representations”.
855 In support of these allegations, the applicant relied upon the evidence to which I have referred briefly at paras 259‑296 above, and much more. I have not rehearsed the whole of the extensive (largely documentary) evidence on which the applicant relied, because central to its utility in the applicant’s case is the allegation that the Vioxx representations were disseminated to pharmacists, medical practitioners and other health care professionals. Quite obviously, only if it were proven that a particular representation was made to, or came to the attention of, a doctor (etc) whose position was relevant to the decision of a particular group member to take, or to continue taking, Vioxx would that representation have the potential to perform service in the applicant’s negligence case. By the concept of “relevance” here I include, of course, both direct and indirect relevance, allowing for the possibility, for example, of a directly relevant doctor having been influenced by more senior members of the profession and the like. However, the mere fact of marketing “messages” in various terms having been formulated within the MSDA marketing department would not be sufficient to provide the evidentiary link that such messages came to the attention of, and influenced, the practitioners who ultimately made the decision, or gave the advice, which led to Vioxx being prescribed for a particular patient.
856 It is also important to make a discrimination between different kinds of promotional material and means of communication. In relation to the “Dear Doctor” and “Dear Pharmacist” letters, I refer to what I have written at para 273 above. They constitute evidence of communications which were specific in the terms in which, and general in the addressees to whom, they were sent. Another class of document was the publication of an entry in a journal or periodical circulating generally in the medical profession, such as the “advertorials”. A publication of that kind would represent an objectively observable communication which may be presumed to have been accessible to the profession as a whole, and, to the extent that it was read by practitioners, would amount to a single communication made in the same terms to each of them. But I could not infer that every such publication was necessarily read by every practitioner. Next in order would be documents which may be assumed to have informed the general approach of sales representatives, most obviously the “detail aids”. However, I could not find that the mere existence of such a document justified the inference that its terms were represented, as such, to every practitioner. The particular use to which such documents were put would depend upon choices made by individual representatives from situation to situation. Next again would be documents the use of which was, seemingly, entirely contingent, most obviously the “objection handling modules”. By definition, the use of these documents depended on a particular doctor, for example, raising some query or the like about Vioxx.
857 These considerations show that it is not possible, at this stage of the case, to find that every document produced by MSDA with an ostensible marketing purpose represented a communication made in the terms indicated to every member of the class of apparent addressees. That is not to say that I reject the applicant’s case that MSDA, having devoted considerable funds to the promotion of Vioxx in Australia, would be unlikely not to have ensured that its marketing message had the intended impact at the level of prescribers and other relevant health care professionals. But an impressionistic conclusion at that level would clearly be insufficient, standing alone, to provide a basis for a positive finding that a particular representation or communication had been made to and perceived by every relevant practitioner in Australia. At that level, I shall have to receive evidence which is specific to the circumstances of the individual group members, as I have in the case of the applicant himself.
858 Likewise, the practical utility of the evidence led by the applicant about MSDA’s marketing campaign and strategy would seem to depend upon the extent to which the message − effectively the contents of documents developed internally to MSDA − was communicated externally in a material context, or possibly because it enabled an inference to be drawn in support of some otherwise contested evidence about such a communication. By the second possibility, I have in mind, for example, that there may be conflicting evidence about what a representative said to a doctor on a particular occasion. It might then be possible to infer that the representative was more likely to have been speaking consistently with some recent internal instruction or memorandum issued by MSDA. However, absent any primary evidence of a representation that would advance the case of a particular group member, it is not easy to see how the court could infer that a certain conversation was had simply on the ground that to have had such a conversation would have been consistent with a the marketing campaign as developed internally.
859 Turning then to the applicant’s own case, it is established that, in their meetings with Dr Dickman, the MSDA representatives emphasised the safety of Vioxx (see paras 717‑719 above). That emphasis was not consistent with the cardiovascular risk signal given by the VIGOR trial, which was known to MSDA at all times when Dr Dickman was prescribing Vioxx to the applicant. Neither, for that matter, was it consistent with MSDA’s own detail aids, which confined the claims made about safety to gastrointestinal safety. I accept the applicant’s case that the emphasis given to safety in those unqualified terms was inconsistent with the proper discharge by MSDA of the duty of care which it owed to the consumers of Vioxx.
860 There is, however, no evidence that any other promotional representation (other than the “Dear Doctor” letters to which I have referred) made by or on behalf of MSDA came to the attention of Dr Dickman (or, for that matter, of Dr Anderson). For example, neither Dr Dickman nor Dr Anderson gave evidence that he queried an MSDA representative in any circumstances that might make any of the objection handling modules relevant to the circumstances of the applicant. Neither of them gave evidence that they read, or had read to them, the contents of a detail aid, or ever saw a “leave‑behind card”.
Causation
861 I have decided the matter of biological causation favourably to the applicant, but I have also held that the proper discharge of the respondents’ duty of care did not require them to withdraw Vioxx from the market sooner than they did. The legally relevant causation questions, therefore, are whether MSDA’s failure to warn, and/or its emphasis upon safety, caused Dr Dickman to prescribe, and to continue to prescribe, Vioxx to the applicant, and caused the applicant to continue to consume it. Before turning to those questions as such, however, I should deal with an aspect of the respondents’ submissions which is appropriately dealt with in this section on causation.
862 The respondents submitted that, regardless of any steps taken or omitted by MSDA, Dr Dickman and the applicant were aware that the consumption of Vioxx was associated with a potential cardiovascular risk. There was, however, nothing in the evidence that would sustain such a conclusion in the case of the applicant himself. As to Dr Dickman, the respondents’ submissions were based upon the evidence to which I have referred in summary at paras 721‑723 above. It is not established, on that evidence, that Dr Dickman had in fact read the October 2001 newsletter of the National Prescribing Service, the June 2003 newsletter from the same source or the October 2003 bulletin published by the Australian Adverse Drug Reaction Advisory Committee. Such evidence on the subject as he was induced to give under cross‑examination was both hypothetical and tentative. I am not satisfied that Dr Dickman was in fact aware, at any time which is presently relevant, of the risk that the consumption of Vioxx might be associated with the occurrence of cardiovascular events, or of the results of the VIGOR trial.
863 On the other hand, I do accept that, at some time between December 2001 and September 2004, Dr Dickman came to be aware of the “ideas encapsulated” in the new paragraph introduced into the Vioxx Product Information in November 2001. He was not aware of those ideas before that month. Further, it has not been established when, within the 33‑month period referred to by cross‑examining counsel, he first became aware of those ideas. Relevantly to the circumstances of the applicant, it has not been established that Dr Dickman became so aware before December 2003. I accept that, in the light of the evidence to which I have referred, there should be some qualification to Dr Dickman’s evidence in chief that he was not aware of an increased cardiovascular risk associated with Vioxx at the time that he prescribed it to the applicant. However, I do not accept that that evidence has been undermined to the extent necessary to justify the conclusion that, at some time before December 2003, Dr Dickman became aware either of an increased cardiovascular risk as such or of the kind of worrisome signal of potential risk that ought to have been notified to him by MSDA.
864 What would have happened had an appropriate warning been given to Dr Dickman? That is a crucial aspect of causation in a negligence case based on failure to warn: see eg Rosenberg v Percival. There is a certain artificiality about the task thus presented to the court in a product liability (as distinct from a professional liability) case, particularly where the product was consumed consistently over a period. However, I consider it is necessary to look at the matter as at the time when the duty first arose, and by reference to a hypothetical warning no more categorical than has been held to be the minimum necessary to discharge the manufacturer’s duty of care in the circumstances obtaining. I have held that a warning should have been given no later than October 2000, but had it been given to Dr Dickman at any time before 10 May 2001 it would presumably have had the same relevance to his decision to prescribe Vioxx to the applicant on that day. As to the terms of the warning, I have held that the terms of the new paragraph in the Vioxx Product Information inserted in November 2001 would have been sufficient.
865 How would Dr Dickman have responded to a warning in those terms, if given to him by correspondence at some time before 10 May 2001? By then, although Vioxx had been on the PBS for about four months only, Dr Dickman’s experience with it had been very positive: see the entries for 30 January, 15 February, 8 March and 20 April 2001 in the list in para 718 above. In the balancing of the considerations that he would have undertaken had he been aware that Vioxx involved an increase in cardiovascular risk (see para 720 above), I consider that this positive experience would have made a contribution of some substance. Further, it seems that Dr Dickman regarded the absolute risk rate of CVT events for patients taking Vioxx as disclosed in VIGOR − 1.67 events per 100 patient‑years − to be low in the circumstances of the applicant, whom he regarded as a “mild to moderate” cardiac risk. With all the problems that the making of a judgment about a hypothetical situation involves, I am persuaded that the most likely response of Dr Dickman to a warning in the terms that I have held to be sufficient would have been to advise the applicant that the increased risk of a cardiovascular event was relatively low. While Dr Dickman would have left the final decision to the applicant, my impression is that he would have placed rather more emphasis upon the benefits of Vioxx and rather less upon the cardiovascular risk.
866 As Dr Dickman made clear in his evidence, the choice facing the applicant was whether to take a coxib or a conventional NSAID. To have left his back pain untreated was not a realistic option. Dr Dickman well understood the sense of the “Cardiovascular Effects” paragraph in the Product Information of November 2001. He understood that, relevantly for a patient who needed a drug for his arthritis, the difference between naproxen and Vioxx was that the former had an antiplatelet action while the latter did not. He is likely to have explained this to the applicant, and also to have pointed out that the other side of the coin was that naproxen would be likely to be associated with a greater risk of gastrointestinal side‑effects.
867 How would the applicant have responded to the kind of advice from Dr Dickman that I have found would most likely have been given? The applicant described himself as a fairly stubborn person who preferred to reach his own conclusions, rather than accepting someone else’s word; and as cautious and safety‑conscious. He said that he was “wary of medication”. While he was in the Navy, he experienced a bad reaction from having omitted to read the label on some migraine tablets that had been prescribed at twice the strength of those that he had previously taken. This taught him a lesson about reading labels and checking carefully what he was taking. He regarded himself as risk‑averse, and would “tend to evaluate the pros and cons before making any important decision”.
868 I refer also to the applicant’s evidence as I have referred to it at paras 737‑741 above. The applicant respected Dr Dickman and placed store by his advice. He was not in terms asked how he would have responded to news that patients on Vioxx in the VIGOR trial experienced CVT events at a rate of 1.67 per 100 patient‑years, but he was asked in chief how he would have reacted to advice that the risk of a heart attack was two, three or four times as high on Vioxx as on naproxen. He said that he would have wanted to know where the figures came from, beyond which he declined to speculate: see para 738 above. Had he asked Dr Dickman where the figures came from, I consider that Dr Dickman would have told him that VIGOR was a trial involving patients with rheumatoid arthritis, that the dose of Vioxx in the trial was double what was then proposed, and, as mentioned above, that naproxen had an antiplatelet action whereas Vioxx did not. As I have said, I consider that Dr Dickman’s advice would have been generally favourable towards Vioxx. There was, additionally, the applicant’s long and unhappy experience with the gastrointestinal side‑effects of NSAIDs, which would have provided him with an incentive to opt for a coxib.
869 The present question is one on which the applicant carries the onus of proof. Having regard to the matters referred to above, I am not persuaded that, had the applicant been advised by Dr Dickman in terms which reflected the warning which would have been a reasonable and sufficient response to the VIGOR results, he would have declined to take Vioxx. He may have done so, but he has not established that such a course should now be regarded as more likely than the course which he did adopt, which was to take Vioxx. Since the applicant’s experience with Vioxx was entirely positive, neither am I persuaded that, if such advice had been given to him at some point between May 2001 and December 2003, he would have discontinued taking Vioxx. Thus I am not satisfied that MSDA’s failure to warn was a contributing cause to the applicant’s consumption of Vioxx or (therefore) to his heart attack in December 2003.
870 I deal next with causation in the context of the MSDA sales representatives’ emphasis upon the safety of Vioxx in their conversations with Dr Dickman (see para 717 above). To what extent did Dr Dickman rely upon that emphasis? Had the representatives said nothing at all about general safety (ie had they confined their promotional talk to the matter of gastrointestinal safety, where MSDA was on surer ground), would he nonetheless have prescribed, and continued prescribing, Vioxx for the applicant?
871 Dr Dickman was an unambiguous supporter of coxibs. He prescribed Vioxx and Celebrex about equally, and considered that it would have been almost negligent of him to have prescribed a conventional NSAID for a patient with a complaint for which a coxib was indicated. In the case of Vioxx, this derived originally from his attendance at the dinner in late 1999 or early 2000 at which the specialist rheumatologist spoke. His experience with Vioxx was universally positive, the applicant himself being an example of a patient for whom Vioxx had brought relief from pain without gastrointestinal side‑effects. He was a very experienced general practitioner who made his own judgments about drugs.
872 Dr Dickman was cross‑examined as follows:
It’s fair to say that you from at least 2000 onwards proceeded on the basis that no prescription medicine was absolutely safe for every patient?---That’s correct.
One couldn't proceed that [way], could you?---No.
[What] you had to do was to learn about the range of effects of the drug, ranging from good through to serious adverse effects or possible serious adverse effects, and then make a judgment about whether it should be prescribed for the particular patient?---Yes.
If a drug representative came to you and said, “Here is a prescription medicine. I can guarantee you that it’s absolutely safe”, you would shake your head and think, “I think that’s an overstatement”, wouldn’t you?---Yes.
Because that doesn’t accord with your experience of prescription medications?---No.
You would think to yourself, “I had better go and have a look at what I can find out about this drug to see what the safety of it is”, wouldn’t you?---Yes.
You certainly wouldn’t just take the rep’s word for it, would you?---No.
Indeed, whenever a rep made a comment about safety of a drug you knew that it had to be placed in the context of all of the information that was available about the drug?---Yes.
These answers were entirely consistent with the impression I had of Dr Dickman generally. In short, I do not think he relied on the emphasis given by MSDA sales representatives as to the safety of Vioxx. Had these statements not been made to him, I am satisfied that he would in any event have prescribed, and continued to prescribe, Vioxx for the applicant.
873 Counsel for the applicant stressed that MSDA’s failure to discharge its duty of care should be recognised for the overall impression created by its acts and omissions generally, rather than by addressing each element of that failure in isolation from each other element (as arguably I have done above). They submitted that the court should consider the impact on general practitioners such as Dr Dickman of MSDA’s failure to warn in an environment in which inappropriately positive marketing messages were being conveyed by sales representatives. I accept those submissions. However, even by reference to a hypothetical situation in which a warning in terms which I have held to be sufficient had been given to Dr Dickman, and in which there were no marketing representations about the “safety” of Vioxx, I am not persuaded that he would have withheld from prescribing Vioxx for the applicant, or that the applicant would have declined to take Vioxx.
874 For the above reasons, I propose to dismiss the applicant’s case in negligence against MSDA.
part viii − the applicant’s case under the trade practices act
Implied repeal
875 I have briefly, and very broadly, introduced the applicant’s claims under the Trade Practices Act at para 17 above. Before turning to those claims as such, I should mention a sweeping, and potentially decisive, submission made by the respondents with respect to all of the provisions of the Trade Practices Act on which the applicant relies. They submitted that these provisions had been implicitly repealed by the TG Act. This contention was the subject of no mention in the final oral submissions made on behalf of the respondents. I suppose it was covered by the safety‑net submission made at the outset by counsel for the respondents that they adopted their written submissions, and ought not to be taken to have abandoned any part of them just because they did not mention them in the course of their oral address. The subject of implied repeal was dealt with in a cursory and, I would have to say, quite unsophisticated, way in the respondents’ written submissions. However, the submission was technically made, and must be dealt with.
876 When two different statutes made by the same legislature deal with the same general subject‑matter, the presumption is that the legislature intended that both provisions should operate: Ferdinands v Commissioner for Public Employment (2006) 225 CLR 130, 138 [18] and 146 [49]. In that case, Gummow and Hayne JJ said that, in deciding whether there was inconsistency (or “contrariety” or “repugnancy”), what was required was “the construction of, and, close attention to, the particular provisions in question”: Ferdinands at 138 [18]. Sadly, the respondents’ submissions overlooked that requirement. They provided no close attention either to the provisions of the Trade Practices Act upon which the applicant sues, or to the provisions of the TG Act which were said to be inconsistent with them. They confined themselves to a reference generally to the provisions of Pt 5‑1, and of Div 3 thereof, in that Act, and drew particular attention to s 42DL(1)(f).
877 Section 42DL(1)(f) of the TG Act provided, in 2004, as follows:
A person must not publish or broadcast an advertisement about therapeutic goods:
…
(f) that refers to goods, or substances or preparations containing goods, included in Schedule 3, 4 or 8 to the Poisons Standard ….
Relevantly to the present circumstances, the “Poisons Standard” was defined to mean –
… the latest edition at the commencement of this Part of the document known as the Standard for the Uniform Scheduling of Drugs and Poisons published by the Australian Health Ministers’ Advisory Council.
The document described as the “Standard for the Uniform Scheduling of Drugs and Poisons” was not in evidence. It was not referred to by any witness. It was not referred to in the submissions of the respondents. On this ground alone, I would have to reject the respondents’ point on implied repeal.
878 I would add that s 42DL(1)(f) of the TG Act was, according to the submissions of the respondents, added to the TG Act only in 2003. Those submissions gave me no assistance as to how I should understand the point about inconsistency to the extent that it was said to render inoperative the provisions of the Trade Practices Act before that amendment.
879 In final oral submissions made on his behalf, the applicant pointed out that Pt 5‑1 of the TG Act (on which the respondents relied) does not apply to advertisements directed exclusively to medical practitioners, pharmacists and other identified health care professionals. There was no response to this submission in the reply submissions made on behalf of the respondents. Neither did the respondents’ written submissions advert to this exclusion. To the extent that the applicant’s allegations under the Trade Practices Act involve the interface between MSDA and medical practitioners or pharmacists, and to the extent that the provisions broadly relied upon by the respondents under the TG Act might otherwise be thought to provide some sustenance for their argument on implied repeal, this provision would appear to be a complete answer.
880 It was also submitted on behalf of the respondents that part of the system implemented by the TG Act involved the regulation of the type of information provided to the public and to prescribers by the power to impose conditions on the approval of Product Information. Section 28 of the TG Act was referred to, and it does, as I have noted elsewhere, contain a power to impose conditions. However, the respondents submitted that this power was used to “require the sponsor to publish the Product Information in an approved form”. No example of the s 28 power being used to impose such a condition was cited and, for my own part, I have found none in the evidence. The s 28 conditions imposed on MSDA in the case of Vioxx were in evidence, and I have referred to a number of them. Nowhere was MSDA required to publish the Product Information. Neither does the TG Act itself − or the regulations made under it – impose such a requirement. As I have indicated, the Act contemplated that there would be “product information”, and the 1994 guidelines required that information to be provided to the TGA, upon the making of an application for registration, in a particular form. It was also a condition of the registration of Vioxx that the Product Information not be changed without the approval of the TGA. However, I can find nothing, either in the Act or elsewhere, which imposed upon MSDA an obligation to publish the Product Information.
881 It will be apparent that I think very little of the respondents’ argument on implied repeal, and I do not propose to occupy further time giving it the content or the attention which the respondents themselves failed to do.
Section 52
882 Section 52(1) of the Trade Practices Act provides as follows:
(1) A corporation shall not, in trade or commerce, engage in conduct that is misleading or deceptive or is likely to mislead or deceive.
883 The applicant’s case under this provision was based first upon what he described as “the Vioxx circumstances” and “the “Vioxx conduct”. He defined the Vioxx circumstances as:
(a) rofecoxib was a prescription only non‑steroidal anti‑inflammatory drug;
(b) [MSDA] marketed in Australia Vioxx tablets to pharmacists, medical practitioners and other health care professionals as a non-steroidal anti‑inflammatory drug for the treatment of arthritis that was safer than other non‑steroidal anti‑inflammatory drugs in that it was less likely to cause adverse gastrointestinal conditions and reactions;
(c) [MSDA] knew that medical practitioners and other health care professionals in Australia would prescribe, and pharmacists in Australia would supply under prescription, Vioxx tablets as a non‑steroidal anti-inflammatory drug for the treatment of arthritis;
(d) [MSDA] knew that persons who were prescribed by a medical practitioner or other health care professional in Australia and obtained from a pharmacist in Australia Vioxx tablets would consume the Vioxx tablets as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis;
(e) [MSDA] knew that many persons who were prescribed by a medical practitioner or other health care professional in Australia and obtained from a pharmacist in Australia Vioxx tablets and who consumed the Vioxx tablets as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis were likely to be elderly and, accordingly, at particular risk of suffering the [pleaded] cardiovascular conditions;
(f) [MSDA] provided to pharmacists, medical practitioners and other health care professionals in Australia product information with respect to Vioxx tablets (“the Vioxx product information”) setting out warnings and precautions with respect to the consumption of rofecoxib or Vioxx tablets and adverse reactions that may be associated with the consumption of rofecoxib or Vioxx tablets;
(g) the Vioxx product information did not contain any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(h) [MSDA] knew or ought reasonably to have known that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions.
He defined the Vioxx conduct as:
(a) in packaging and labelling Vioxx tablets, [MSDA] failed to provide any adequate information, advice or warning on packets of Vioxx tablets or the labelling thereon to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions; and/or
(b) in marketing in Australia Vioxx tablets, [MSDA] failed to provide pharmacists, medical practitioners and other health care professionals to whom Vioxx tablets were marketed in Australia any adequate, information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions; and/or
(c) in distributing and/or supplying for sale in Australia Vioxx tablets, [MSDA] failed to provide to pharmacists, medical practitioners or other health care professionals to whom Vioxx tablets were distributed and/or supplied any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions.
He alleged that the Vioxx conduct was engaged in in trade and commerce, and that it was misleading or deceptive, or likely to mislead or to deceive. As is apparent, here the applicant’s case is that MSDA’s conduct was constituted by its failure to do something.
884 The other aspect of the applicant’s s 52 case is based upon the “Vioxx representations” to which I have referred in para 854 above. He alleged that those representations (and the marketing campaign out of which they arose) amounted to conduct in trade and commerce which was misleading or deceptive, or likely to mislead or to deceive, in that the consumption of Vioxx materially increased the risk of suffering the pleaded cardiovascular conditions, and in that Vioxx was not safer in that respect than other NSAIDs.
885 The respondents submitted that the conduct upon which the applicant relied for the purpose of s 52 was not in trade or commerce. They relied upon Concrete Constructions (NSW) Pty Ltd v Nelson (1990) 169 CLR 594, 604:
What the section is concerned with is the conduct of a corporation towards persons, be they consumers or not, with whom it (or those whose interests it represents or is seeking to promote) has or may have dealings in the course of those activities or transactions which, of their nature, bear a trading or commercial character.
The respondents then submitted that the conduct of which the applicant complained was “conduct which occurred in relation the prescription of Vioxx to him (and to the Group Members)” and “involved reliance by MSDA on the [Product Information] as the primary vehicle by which information and warnings about Vioxx were conveyed to medical practitioners”. The first aspect of this defence is correct so far as it goes, but the second, to the extent that it makes any clear proposition about the applicant’s case, is not. It was no part of that case to suggest that the Product Information was conduct in trade or commerce, or was misleading. Rather, the existence of the Product Information, lacking an adequate warning as it did, was said to provide the “circumstances” against which MSDA’s failure to warn should be understood. Or so the applicant alleged.
886 In its Defence, MSDA alleged that it packaged and made available Vioxx tablets for use, on the prescription of permitted prescribers; that it knew that permitted prescribers would prescribe Vioxx tablets and pharmacists in Australia would sell Vioxx tablets under prescription; and that it knew that persons who were prescribed, and who purchased, Vioxx tablets would consume them in accordance with the instructions of their permitted prescribers. These allegations are, relevantly to the present point, tendentiously expressed. They turn a Nelsonian eye to the undoubted fact that Vioxx was sold by MSDA to pharmacists (either directly or through wholesalers – see eg the “Dear Pharmacist” letters to which I have referred at paras 228 and 270 above) by way of conventional (though subsidised) wholesale exchange. The details of that transaction were not the subject of evidence, doubtless because the fact was so obvious as not to require emphasis. MSDA did, of course, operate in a very specialised industry, in which the products it made could be sold only by way of prescription. Thus the end consumer was required to hold a prescription which gave the supplying pharmacist the authority to make the sale. This meant that MSDA’s promotional activities to a large extent were directed to the prescribers. As Ms Dobson said of the MSDA marketing team:
The role of MSDA’s marketing team is to make prescribers aware of the features of its prescription medicines so that they will prescribe them to the broadest group of suitable patients. This is good for patients and good for MSDA, which is a for‑profit company. This was our objective with Vioxx.
Within this specialised framework, MSDA was in the conventional position of a company that manufactured a product for supply to retailers in the expectation that those retailers would sell the product to consumers in need of it.
887 Returning to the Product Information, the respondents submitted that it was “not provided in trade or commerce”. Whether that submission is well‑founded depends not on the content (or original purpose) of the Product Information, but upon the use to which it was relevantly put. In other words, it depends on what one means by “provided”. I am disposed to think that the respondents may be correct to the extent that they submitted that the provision of the Product Information to the TGA for approval was not a transaction of a trading or commercial character. But I do not need to decide that point, since the applicant does not rely upon any such transaction. On the other hand, the evidence is that every time an MSDA sales representative called upon a medical practitioner, he or she would have a copy of the Product Information for every drug then being promoted. The purpose, quite clearly, was to provide the representative with an approved, accurate, source of information to be used at his or her discretion for the purpose of answering questions that might be put by the practitioner. If the Product Information were thus used in the course of what counsel for the respondents described (in his cross‑examination of Dr Dickman) as a “sales pitch”, manifestly there would be conduct in trade or commerce of which the Product Information was an element.
888 It does not matter that the MSDA sales representatives did not themselves take orders, or transact any sales. As the High Court said in Concrete Constructions (immediately after the passage referred to in para 885 above) (169 CLR at 604):
Such conduct includes, of course, promotional activities in relation to, or for the purposes of, the supply of goods or services to actual or potential consumers, be they identified persons or merely an unidentifiable section of the public.
And what I have said about sales representatives applies to the whole range of promotional interactions, or potential interactions, between MSDA and prescribers. A “Dear Doctor” letter, for example, the purpose of which was to promote the prescription of Vioxx, should be regarded as conduct in trade or commerce
889 The respondents relied on R v Small Claims Tribunal; ex parte Gibson [1973] Qd R 490, 493 in support of the proposition that the medical profession did not prescribe medicines in trade or commerce. It was, however, no part of the applicant’s case to contend that it did so. That judgment of the Queensland Full Court was concerned to make the traditional distinction between engaging in trade or commerce and carrying on a profession, in which respect see also Holman v Deol [1979] 1 NSWLR 640. But that is not this case. In no sense was MSDA engaged in a professional, as distinct from a trading, activity.
890 The respondents also submitted that so much of MSDA’s activities as were constituted by the (internal) development of its marketing campaign did not amount to conduct “in trade or commerce” because such activities did not occur “in the marketplace”. Such a proposition is too sweeping. It is clear from Concrete Constructions that things said and done (and, for that matter, omissions) internally to a trading organisation may, in appropriate circumstances, amount to conduct in trade or commerce within the meaning of s 52. All will depend on context. However, the limits of such a proposition do not need to be tested in the present case. To the extent that it was based on what was said to be MSDA’s marketing campaign for Vioxx, the applicant’s case required that the “Vioxx representations” be disseminated to pharmacists, medical practitioners and other health care professionals. His s 52 case did not involve the contention that, merely by internally developing that campaign or an associated strategy, MSDA would contravene the section. In one aspect of the way the applicant developed his case, he did submit that an inference of external dissemination should be drawn from the internal development of the marketing campaign. That is an evidentiary question to which I shall turn, but in no sense was the applicant contending that the internal development of the campaign was, as such, the conduct in trade or commerce on which he relied.
891 It was likewise with so much of the s 52 case as was based on an allegation in effect, of misleading conduct by failure to warn. The “Vioxx conduct” (see para 9 above) essentially involved failures to communicate. The applicant was here concerned with the interface between MSDA and others, be they consumers, pharmacists, doctors or other health care professionals.
892 I turn then to the primary elements of the applicant’s case under s 52. Because of the degree of overlap between the applicant’s case in negligence and his case under the statute, I shall assume a familiarity with what I have written in Part VII above, and shall attempt to keep repetition to a minimum.
893 The applicant’s case of misleading conduct by commission was based, as I have said, on an allegation that the “Vioxx representations” were disseminated to doctors and others. In the case of any particular group member, however, the question will be whether MSDA engaged in such conduct in a way that was relevant to him or her. Save for the circumstances of the applicant himself, I could not make a finding that MSDA did engage in such conduct purely on the evidence led to date. Assuming a paradigm in which the representations involved in the conduct were made to, and relied upon by, the prescribing practitioner of a particular group member, such a finding will need to await the calling of evidence on that subject.
894 In the case of the applicant, Dr Dickman could not recall the specifics of any particular interaction with MSDA sales representatives, and he gave no evidence about ever having seen any written promotional material for Vioxx.
895 Of the “Vioxx representations” (see para 854 above), I could not find that those referred to in (c)‑(f) were ever made to Dr Dickman. The broad accuracy of that referred to in (a) is not seriously controversial, and I do not understand the respondents to have resisted the suggestion that it was made to Dr Dickman. It is the representation referred to in (b) − that Vioxx has an excellent overall safety profile compared with other NSAIDs − that corresponds broadly with Dr Dickman’s evidence that the MSDA sales representatives’ emphasis was primarily on Vioxx’s safety. For reasons explained elsewhere, I accept that evidence. That message was, in my view, misleading. Even against the scientific knowledge known to MSDA before September 2004, it would have been misleadingly categorical to say that Vioxx was safe. I would hold, therefore, that MSDA contravened s 52 when its sales representatives conveyed this broad safety message to Dr Dickman.
896 I turn next to so much of the applicant’s case as involved the proposition that the court should infer, from the size, nature, sophistication and cost of the internal marketing campaign developed by MSDA, that the “Vioxx representations” were in fact made externally. This inference was said to be open because (1) it was MSDA’s purpose to disseminate those representations; (2) MSDA spent very large sums on its marketing campaign; and (3) the campaign was successful, in that, soon after its launch, Vioxx “rapidly penetrated the competitive target market”. These very high‑level submissions do not, however, come to grips with the very low‑level evidentiary task which confronted the applicant: to prove, either by direct evidence or by necessary inference, what was said or otherwise communicated by MSDA about Vioxx, to whom, when, and in what context. The “Vioxx representations” themselves are in the nature of a catalogue of statements (or paraphrases of statements) to be found in various documents discovered by MSDA. In his final submissions, however, the applicant did not link the evidence as called to each of these representations. In the circumstances, he has left too many dots unjoined in the proof of what representation was made to whom, and when. On the evidence in the case, I could not hold that a particular representation about Vioxx was made to every general practitioner in Australia.
897 The problem is presented in sharp relief by the evidence of the general practitioners whom the applicant did call, Drs Dickman, Anderson and Lynch. Save with respect to the “safety” emphasis of which Dr Dickman gave evidence, none of these doctors said that the Vioxx representations had been made to him. The best evidence of the making of the Vioxx representations would have come from these witnesses, based on their own experiences. Had there been some infirmity of recollection at the point of detail, the timing and content of the marketing campaign as developed internally might have provided some corroboration and, possibly, some assistance by way of inference. But, these doctors having given no evidence about the subject at all, it would be going too far to infer, merely from what MSDA did internally, that they were all most probably addressees of the “Vioxx representations”.
898 The applicant contended also that MSDA effectively marketed Vioxx by having opinion leaders in the profession, such as specialists, talk up the benefits of the drug at conferences, dinners and the like. The only evidence of a prescribing practitioner having been affected by such representatives, however, was of Dr Dickman’s attendance at a dinner addressed by a rheumatologist in late 1999 or early 2000. This was, of course, before the VIGOR results were available. However that may be, I could not hold MSDA accountable for what the rheumatologist said. I also note Dr Dickman’s oral evidence that what he learnt at that dinner was that Vioxx had the same or a similar overall effectiveness and safety profile to NSAIDs, but that it had a particular gastrointestinal safety benefit.
899 Turning next to the applicant’s case on misleading and deceptive conduct by omission, under the Trade Practices Act one may engage in conduct by refraining (other than inadvertently) from doing an act: s 4(2)(a) and (c). To the extent that MSDA’s failure to warn amounted to refraining from warning, it was not submitted by the respondents that it was inadvertent. From the evidence in the case, it is clear that the relationship between the consumption of Vioxx and adverse CVT events, and the medical profession’s perception of that relationship, was very much part of the conscious thoughts of those responsible for marketing and selling Vioxx.
900 As with their response to the applicant’s negligence case on failure to warn, the respondents never really came to grips with the corresponding aspect of his case under s 52. They tended to deal with the problem of “silence” (as they described it) in terms of generalities. In opening, their counsel said:
Silence is a circumstance on the authorities like any other which needs to be considered in the context in which it occurs and … in our respectful submission will ultimately prove to be an individual matter.
In their final written submissions, they said that courts had “expressed a reluctance to set down exhaustive principles by which cases involving allegedly misleading or deceptive conduct by silence may be characterised”. They adverted to what was said by Black CJ in Demagogue Pty Ltd v Ramensky (1992) 39 FCR 31, 32:
Silence is to be assessed as a circumstance like any other. To say this is certainly not to impose any general duty of disclosure; the question is simply whether, having regard to all the relevant circumstances, there has been conduct that is misleading or deceptive or that is likely to mislead or deceive. To speak of “mere silence” or of a duty of disclosure can divert attention from that primary question. Although “mere silence” is a convenient way of describing some fact situations, there is in truth no such thing as “mere silence” because the significance of silence always falls to be considered in the context in which it occurs. That context may or may not include facts giving rise to a reasonable expectation, in the circumstances of the case, that if particular matters exist they will be disclosed.
See also 41 (Gummow J). The respondents referred also to Johnson Tiles Pty Ltd v Esso Australia Pty Ltd (2000) 104 FCR 564, 591−592 [67] and to E v Australian Red Cross Society (1991) 27 FCR 310, 350. These were, with respect to the respondents, submissions at a very high level which engaged little with the actual facts of the present case.
901 Where the respondents did touch upon the facts of the case was in their submission that the applicant’s allegation of silence “is made despite the PI, which was the principle [sic] vehicle by which precautions and warnings were conveyed, having been approved by the TGA”. This submission does, of course, beg the question, and it does so in two respects. First, it assumes that use of the Product Information as the “principal vehicle” by which precautions and warnings were conveyed legitimises the absence of other forms of communication for such purposes; and secondly, it assumes either that the Product Information at all times said everything that needed to be said by way of warning or, to the extent that it did not, that it was not open (or at least not required as a matter of reasonable response to the emerging science) for MSDA to go beyond it. Each of these propositions is, of course, highly contentious.
902 However that may be, the respondents’ submission leaves no doubt about their position in the litigation. It was that all relevant medical practitioners might be assumed to have ready and convenient access to the Product Information. They cross‑examined Drs Lynch and Dickman by reference to such a proposition. That makes the “Vioxx circumstances” alleged by the applicant significant, particularly the following:
(f) [MSDA] provided to pharmacists, medical practitioners and other health care professionals in Australia product information with respect to Vioxx tablets (“the Vioxx product information”) setting out warnings and precautions with respect to the consumption of rofecoxib or Vioxx tablets and adverse reactions that may be associated with the consumption of rofecoxib or Vioxx tablets;
(g) the Vioxx product information did not contain any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions …
MSDA denied that it “provided” the Product Information to pharmacists etc, alleging instead that it “made Product Information … available”, which I consider to be a more accurate statement of the situation. Subject to that qualification, the respondents’ position clearly was that doctors and pharmacists might be expected to have regular reference to the Product Information as an accurate, complete and TGA‑approved source of information about Vioxx, including its potential risks and side‑effects, to the extent that there were any.
903 At all times until November 2001, the Vioxx Product Information lacked any warning which conveyed the results of the VIGOR trial. Was this misleading conduct by silence? I think it was. A practitioner reading the Product Information would, in my view, reasonably have assumed that what was said about the cardiovascular side‑effects was what MSDA knew on the subject. It was said, for instance, that myocardial infarction had been observed in less that 0.1% of patients taking Vioxx in osteoarthritis studies. That was literally true, but in mid‑2000 and thereafter it was misleading since MSDA was then in possession of the VIGOR results which contained a figure of 0.5% based on patient‑years. There were, of course, all manner of qualifications that could, and perhaps should, have been made with respect to a direct comparison between the osteoarthritis studies data and the VIGOR data (eg different patient population, different dose), but these would have been understood by practitioners and given such weight as was appropriate in the making of their professional judgments. As the respondents stressed, the Product Information was written for a professional readership. I also note that, from very soon after the release of the VIGOR results, MSDA took the view that some amendments were required thereby, notwithstanding that Vioxx was then registered in relation to osteoarthritis only (and notwithstanding also that MSDA itself initially proposed that the gastrointestinal results only be the subject of an amendment to the Product Information). However the matter is looked at, to have given medical practitioners no advice of the results of the VIGOR trial when the unamended Product Information was at large amounted to misleading conduct.
904 I stress that it is no part of my reasoning to hold that the Product Information was itself misleading. The applicant did not so contend. Rather, the misleading conduct was constituted by the failure to draw to the attention of doctors and other health care professionals the cardiovascular risk message that emerged from VIGOR. The respondents criticised the applicant for not having articulated the terms that any such advice would embody. Not having done anything, however, they are in no position to make that criticism. Ultimately, they did come up with a form of advice which I have held to be sufficient for the purposes of the applicant’s negligence case. But before that occurred, MSDA’s failure to provide any qualification to what was then contained in the Product Information did, for the reasons I have given, amount to misleading conduct.
905 For the same reasons, a different conclusion must be reached once the Product Information was amended in November 2001. No longer could it be said that the existence of the original Product Information made MSDA’s failure to warn misleading. This is a different point from my earlier conclusion that merely to cause an amended Product Information to be approved by the TGA was not a sufficient warning to doctors. Here the question is not whether MSDA warned: it is whether it misled. It is quite possible to refrain from warning without misleading. Given that the amended Product Information was at large from November 2001, and that it fairly − albeit compendiously − referred to the cardiovascular results of VIGOR, it could no longer, in my view, be said that merely by remaining silent MSDA had engaged in misleading conduct. It is true that the new paragraph in the “Precautions” section did not advert to the myocardial infarction figure from VIGOR as such, but it did refer to the CVT rate as a whole. The 0.5% myocardial infarction figure was part of the CVT figure of 1.67%. The citation of the latter was, in my view, sufficient to absolve MSDA of the charge, made by the applicant, that its failure to advise doctors directly of the myocardial infarction risk signal given by VIGOR was misleading.
906 I would add two riders to what I have just written. The first is to emphasize that I have held that it was misleading for MSDA not to have made any communication to doctors (etc) of the cardiovascular results of the VIGOR trial while the pre‑November 2001 Product Information was abroad. I do not hold as a matter of fact that it did not do so, save in the case of Dr Dickman. Although there is no evidence of any such communication, I allow for the possibility that the practitioner treating a group member other than the applicant might have been addressed by a representative, or otherwise have received a communication from MSDA, in terms which would have rendered MSDA’s conduct, viewed in the overall context, non‑misleading. The second rider is to make clear, lest there should otherwise be any misunderstanding, that I do not reach the conclusion expressed in the previous paragraph with respect to the positively misleading representations that Vioxx was safe. I do not consider that the amendment made to the Product Information in November 2001 rendered such representations − to the extent that they were made thereafter to Dr Dickman − non‑misleading.
907 That brings me to s 82 of the Trade Practices Act. By that provision, the applicant would be entitled to recover from MSDA the amount of any loss or damage which he suffered “by” the conduct which was in contravention of s 52. It was submitted on his behalf that the consumption of Vioxx caused (or contributed to) his heart attack, and that the misleading conduct of MSDA caused him to be prescribed Vioxx by Dr Dickman. The respondents accepted that, in a case under ss 52 and 82, it was not necessary for the injured party to have directly relied on the presumptively misleading representation but submitted that the evidence in the present case did not sustain the conclusion that Dr Dickman relied on any misleading representation (either active or silent) by MSDA in his decision to prescribe Vioxx to the applicant.
908 With respect to the period before November 2001, each misleading element of MSDA’s conduct – its broad “safety” message and its failure to report the cardiovascular risk signal given by the VIGOR results – was part of the environment to which Dr Dickman was exposed. Were it necessary to do so, I would reach the same conclusion about this conduct as I did in Part VII above with respect to causation in the applicant’s negligence case. That is to say, I would not be satisfied that, had MSDA’s conduct not contained those elements, or had it contained one only of them, Dr Dickman’s prescribing decisions would have been any different. I would not be satisfied that, in prescribing Vioxx to the applicant before November 2001, Dr Dickman relied either on the broad “safety” message conveyed by MSDA sales representatives or on MSDA’s omission to report the VIGOR risk signal.
909 As it happens, however, I could not be satisfied that there was a biological association between the consumption of Vioxx in and before November 2001 and the applicant’s heart attack in December 2003. That is to say, while I have found in Part VI above that the consumption of Vioxx biologically contributed to the applicant’s heart attack, that finding relied on an earlier finding that the applicant consumed Vioxx more or less continuously in the weeks and months leading up to his heart attack. It follows that it is to the prescribing decisions of Dr Dickman well subsequent to November 2001 that regard should be had for the purposes of the question whether the misleading conduct of MSDA contributed to the applicant’s heart attack in December 2003.
910 The evidence before the court shows that Dr Dickman prescribed Vioxx for the applicant on 10 April, 19 July and 20 December 2002 and 2 December 2003, and that Dr Anderson did so on 30 May 2003. Over the whole of the period when these decisions were being made, MSDA was not engaging in misleading conduct by omission. Its only misleading conduct was constituted by the broad “safety” message conveyed to Dr Dickman by its sales representatives. For reasons given at para 872 above, I am not persuaded that Dr Dickman relied on that message in the decisions he made to prescribe Vioxx for the applicant.
911 It follows that the applicant’s claim for damages under s 82 of the Trade Practices Act with respect to MSDA’s conduct in contravention of s 52 thereof must be dismissed.
Section 75AD
912 Section 75AD of the Trade Practices Act relevantly provides as follows:
If:
(a) a corporation, in trade or commerce, supplies goods manufactured by it; and
(b) they have a defect; and
(c) because of the defect, an individual suffers injuries;
then:
(d) the corporation is liable to compensate the individual for the amount of the individual’s loss suffered as a result of the injuries; and
(e) the individual may recover that amount by action against the corporation;
Under this provision, it should be noted that the defect must exist in the particular goods which presumptively caused injury to the individual. The section is not concerned with the question whether other or similar goods ─ even goods which, as a matter of branding or commercial practice, are “the same” ─ have or had a defect. Thus, although it may be accepted that the Vioxx tablets consumed by all of the group members in the present case were, as a matter of composition, identical (subject, of course, to variations in strength), as will be apparent, the applicant’s allegations of defect travel beyond the matter of composition into areas in which it cannot be assumed that the circumstances which were relevant to himself were also present and relevant for all group members.
913 The “circumstances” to which I refer are the concern of s 75AC of the Trade Practices Act. Subsections (1) and (2) of s 75AC provide as follows:
(1) For the purposes of this Part, goods have a defect if their safety is not such as persons generally are entitled to expect.
(2) In determining the extent of the safety of goods, regard is to be given to all relevant circumstances including:
(a) the manner in which, and the purposes for which, they have been marketed; and
(b) their packaging; and
(c) the use of any mark in relation to them; and
(d) any instructions for, or warnings with respect to, doing, or refraining from doing, anything with or in relation to them; and
(e) what might reasonably be expected to be done with or in relation to them; and
(f) the time when they were supplied by their manufacturer.
These provisions posit two questions for the court: (1) what is the safety of the goods; and (2) is that safety such as persons generally are entitled to expect? It is the first question with which subs (2) is concerned. The second question is to be addressed by an objective approach, rather than, for example, by inquiring what level of safety the relevant individual did in fact expect: Carey‑Hazell at [186].
914 The circumstances to which the applicant pointed for the purposes of s 75AC(2) were the following:
(a) rofecoxib was a prescription only non‑steroidal anti‑inflammatory drug;
(b) [MSDA] marketed in Australia Vioxx tablets to pharmacists, medical practitioners and other health care professionals as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis that was safer than other non‑steroidal anti‑inflammatory drugs in that it was less likely to cause adverse gastrointestinal conditions and reactions;
(c) it was reasonable to expect that medical practitioners and other health care professionals would prescribe and pharmacists would supply under prescription Vioxx tablets as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis;
(d) it was reasonable to expect that persons who were prescribed by a medical practitioner or other health care professional and obtained from a pharmacist Vioxx tablets would consume the Vioxx tablets as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis;
(e) it was reasonable to expect that many persons who were prescribed by a medical practitioner or other health care professional and obtained from a pharmacist Vioxx tablets and who consumed the Vioxx tablets as a non‑steroidal anti‑inflammatory drug for the treatment of arthritis were likely to be elderly and, accordingly, at particular risk of suffering the [pleaded] cardiovascular conditions;
(f) in marketing, distributing and/or supplying for sale in Australia Vioxx tablets [MSDA] failed to provide pharmacists, medical practitioners and other health care professionals any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(g) the packaging and labelling of Vioxx tablets did not contain or carry any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions;
(h) the Vioxx product information did not contain any adequate information, advice or warning to the effect that the consumption of rofecoxib or Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions ….
915 Items (a)–(d) on the applicant’s list are relatively uncontroversial, and I would have little hesitation in finding that they did exist as circumstances in the present case. Item (e) is not so clear‑cut, but, particularly with respect to the period during which Vioxx was indicated only for osteoarthritis, it finds support in the evidence. Criticising the design of the VIGOR trial, Prof Cleland said that the demographic most likely to take Vioxx was that of elderly patients who tended to have greater pre‑existing cardiovascular risks. He was cross‑examined on that evidence, but from a standpoint that accepted that the elderly were at greater risk of cardiovascular disease. In discussing the Merck Alzheimer’s disease studies, Prof Vaughan referred to the elderly as “a population that was at higher risk for cardiovascular disease”. Prof Celemajer said that “the major risk factor for atherosclerosis bar none is increasing age”. As to the typical profile of osteoarthritis patients, counsel for the respondents confirmed my impression that (in his words) osteoarthritis was “a wear and tear type disease [which] will affect people as they get older”.
916 Items (f)‑(h) on the applicant’s list assume, without alleging in terms, the existence of a further circumstance that was alleged by the applicant elsewhere in his pleading: that the consumption of Vioxx did materially increase the risk of suffering the pleaded cardiovascular conditions. I have rejected that allegation for all of those conditions save myocardial infarction. In the case of that condition, I have held that, across a population, the consumption of Vioxx did involve an increase in risk, but that the extent of the risk in a particular case would depend on the conditions presumptively existing in the patient’s vasculature. For a patient in a reasonably advanced state of atherosclerosis, the risk was more pronounced. Whether there was a risk in the case of a particular patient, and how troubling that risk was, were matters which involved a professional judgment by his or her practitioner.
917 Absent the provision of any information, advice or warning, I consider that the risk to which I have referred made the safety of Vioxx less than what persons generally were entitled to expect. Indeed, the withdrawal of Vioxx from the market on 30 September 2004 implied as much. The respondents stressed, and I accept, that many prescription drugs have side‑effects, sometimes quite serious ones. Conventional NSAIDs, with their gastrointestinal issues, are a presently conspicuous example. I could not hold, therefore, that persons generally are entitled to expect that all drugs will be free of side‑effects. However, (and this reflects the broad position adopted by both sides in this case) the ingestion of prescription drugs occurs only after the making of a professional judgment by a medical practitioner, informed by his or her diagnosis of the condition of the patient concerned. Persons generally are, in my view, entitled to expect that, to the extent that a drug is known or believed to have side‑effects, or to carry the potential for side‑effects (particularly of a serious nature), practitioners will, in whatever terms, and by whatever means, are appropriate, be furnished by the drug supplier with information or warnings sufficient to permit a balanced, cautious and informed judgment to be made.
918 As I have held elsewhere, nothing in the nature of a warning, and no information about the cardiovascular results of the VIGOR trial, were communicated by MSDA to Dr Dickman before at least November 2001. Until then, therefore, the safety of Vioxx (as purchased and consumed by the applicant) was not such as persons generally were entitled to expect.
919 Did the safety of Vioxx become such as persons generally were entitled to expect upon the amendment of the Product Information in November 2001? I consider that the existence thereafter of the Product Information in its amended form was an instruction or warning within the meaning of para (d) of s 75AC(2), but the question remains whether, having regard to that and all other relevant circumstances, the safety of Vioxx was not such as persons generally were entitled to expect.
920 In October 2000, MSDA had chosen to correspond with doctors generally in terms which included advice about various side‑effects which might be associated with the consumption of Vioxx, and various precautions which ought to be observed in that regard. An aspect of that advice was that, in certain osteoarthritis studies, the myocardial infarction rate amongst those taking Vioxx had been less than 0.1%. At all times that Vioxx was on the market in Australia, MSDA had available information to the effect that, in the VIGOR trial, the myocardial infarction rate had been 0.5%. Although there may have been a number of legitimate bases upon which MSDA might have proposed that this level of risk was not generally applicable, or was not applicable to osteoarthritis patients, I consider that persons generally were entitled to expect that an advice or a warning that conveyed the outcome of the VIGOR trial would have been communicated to medical practitioners by a means no less likely to capture their attention than the means employed in October 2000.
921 For the above reasons, I am of the view that the mere amendment of the Vioxx Product Information in November 2001 was, in the context of all the relevant circumstances, insufficient to make the safety of Vioxx such as persons generally were entitled to expect. It follows that, when (after that month) MSDA supplied the Vioxx tablets that were ultimately consumed by the applicant, those tablets had a defect within the meaning of s 75AD of the Trade Practices Act.
922 MSDA advances two defences to the applicant’s s 75AD case under s 75AK(1) of the Trade Practices Act. Relevantly to those defences, the subsection provides as follows:
(1) In a liability action, it is a defence if it is established that:
…
(b) [the goods] had that defect only because there was compliance with a mandatory standard for them; or
(c) the state of scientific or technical knowledge at the time when [the goods] were supplied by their actual manufacturer was not such as to enable that defect to be discovered; or…
MSDA relies upon what it describes as the “mandatory standards defence” (para (b)) and upon the “state of the art defence” (para (c)).
923 In s 75AA of the Trade Practices Act, the term “mandatory standard” in relation to goods, is defined as a standard ─
(a) for the goods or anything relating to the goods; and
(b) that, under a law of the Commonwealth, a State or a Territory, must be complied with when the goods are supplied by their manufacturer, being a law creating an offence or liability where there is such non‑compliance;
but does not include a standard which may be complied with by meeting a higher standard.
MSDA submitted that the conditions of the registration of Vioxx on the ARTG did not permit it to manufacture Vioxx outside “the approved specifications”, and did not permit it to add information to the Product Information, except in accordance with the processes for amendment established by the TGA. It submitted that the chemical composition of Vioxx, and the form of the Product Information, existed by reason of its compliance with the conditions imposed by the TGA. It submitted that, where the TGA asked it to include a warning in relation to cardiovascular events in the Product Information, and where there were discussions between it and the TGA in relation to the form of words to be used in that warning, any defect in Vioxx existed only because of compliance with a mandatory standard within the meaning of s 75AA.
924 It will be apparent from what I have written elsewhere in these reasons that I consider these submissions to be quite misconceived. Reading s 75AK(1)(b) with ss 75AC and 75AD, the question is whether the safety of Vioxx was not what persons generally were entitled to expect only because of compliance with a mandatory standard as defined in s 75AA. I have held that the safety of Vioxx was less than what persons generally were entitled to expect because, as a matter of composition, the consumption of Vioxx had the potential to increase the risk of suffering a myocardial infarction, in circumstances which included the absence of any relevant information or warning communicated to the applicant’s doctor. It was not because of a mandatory standard that the composition of Vioxx was as it was. Nor was it because of such a standard that Dr Dickman was not warned. It may have been because of a mandatory standard that MSDA could not change the composition of Vioxx without amending the Product Information, and that the Product Information could not be amended without the approval the TGA. But those circumstances were not what made the safety of Vioxx less than persons generally were entitled to expect. Neither did they have any relevance to the failure of MSDA to warn Dr Dickman, even without amending the Product Information. As I have held, the existence of the Product Information (whether in its amended or unamended form) provided no impediment to MSDA giving such a warning to Dr Dickman or to any other general practitioner.
925 For these reasons, I reject MSDA’s reliance on s 75AK(1)(b) of the Trade Practices Act.
926 With respect to the state of the art defence advanced by MSDA, it submitted that the defect alleged to have existed in Vioxx was that “Vioxx tablets materially increased the risk of suffering the [pleaded] cardiovascular conditions”. MSDA submitted that, while it was not in dispute that there was an hypothesis to this effect during the whole of the period that Vioxx was on the market in Australia, the totality of the scientific evidence did not take the matter further, and “it never rose to the level of scientific knowledge required to enable a defect to be discovered during the relevant period.” In Part V of these reasons, I have concluded that it was not until the unblinding of Merck to the cardiovascular data from the APPROVe study in September 2004 that the respondents knew or ought to have known that the consumption of Vioxx increased the risk of the occurrence of cardiovascular events. To that extent, the respondents’ submission is well‑founded. However, the present question is whether that is one and the same thing as concluding that the defect which I have identified could not have been discovered by reference to the state of scientific or technical knowledge at the time.
927 At the scientific or technical level as such, I would hold that the defect could not have been so discovered. The defect, of course, is the inadequate safety of the goods themselves. Vioxx was unsafe in that sense because it increased the risk of myocardial infarction. However, it was not until September 2004 that that increase in risk could be “discovered” in the sense of established at the scientific level. Merck was at the forefront of research in this regard (understandably, since rofecoxib was its own molecule). Merck’s own knowledge was the state of scientific knowledge to which s 75AK(1)(c) refers. I consider that my conclusion in Part V of these reasons carries the consequence that it was not until September 2004 that the state of scientific knowledge was such as to enable the discovery of the fact that the consumption of Vioxx increased the risk of myocardial infarction.
928 Does that conclusion take the respondents the full distance they need to travel under s 75AK(1)(c)? I ask that question because, on one view at least, by the terms of s 75AC a defect is a situation rather than a particular aspect of the composition of the goods in question. And it is a situation the existence of which must be determined as a matter of judgment only after consideration of all relevant circumstances. In the present case, I have effectively held that persons generally were entitled to expect that MSDA would have given to medical practitioners a warning (etc) which would have conveyed some idea of the signal of risk yielded by the VIGOR trial. The state of scientific knowledge was such as would have enabled such a warning to be given. It was such as enabled MSDA to know of that element of the situational defect as was constituted by the risk signal coming from VIGOR.
929 I do not, however, think that the relevant provisions of Pt VA of the Trade Practices Act should be so construed. Section 75AK(1)(c) contemplates the existence of a defect capable of being discovered by reference to the current state of scientific or technical knowledge. It is not concerned with the kind of contextual circumstances referred to in s 75AC(2). My conclusion that the respondents ought to have acted consistently with the cardiovascular risk signal yielded by VIGOR is in the nature of a judgment as to how persons generally were entitled to expect that MSDA would act. By all means it informed my determination that there was a defect, but the content of persons’ expectations did not constitute the defect itself. The defect was something inherent in Vioxx as a matter of composition. I consider that the intent of s 75AK(1)(c) is that if that defect could not be discovered according to the state of scientific or technical knowledge, the defence should be available, notwithstanding that enough was suspected about the product to activate an implied obligation to give warnings of the kind mentioned in s 75AC(2)(d).
930 For the above reasons, I propose to uphold MSDA’s defence under s 75AK(1)(c) of the Trade Practices Act.
Section 74B
931 Relevantly to the present case, s 74B of the Trade Practices Act provides as follows:
(1) Where:
(a) a corporation, in trade or commerce, supplies goods manufactured by the corporation to another person who acquires the goods for re‑supply;
(b) a person (whether or not the person who acquired the goods from the corporation) supplies the goods (otherwise than by way of sale by auction) to a consumer;
(c) the goods are acquired by the consumer for a particular purpose that was, expressly or by implication, made known to the corporation, either directly, or through the person from whom the consumer acquired the goods or a person by whom any antecedent negotiations in connexion with the acquisition of the goods were conducted;
(d) the goods are not reasonably fit for that purpose, whether or not that is a purpose for which such goods are commonly supplied; and
(e) the consumer or a person who acquires the goods from, or derives title to the goods through or under, the consumer suffers loss or damage by reason that the goods are not reasonably fit for that purpose;
the corporation is liable to compensate the consumer or that other person for the loss or damage and the consumer or that other person may recover the amount of the compensation by action against the corporation in a court of competent jurisdiction.
(2) Subsection (1) does not apply:
(a) …
(b) where the circumstances show that the consumer did not rely, or that it was unreasonable for the consumer to rely, on the skill or judgment of the corporation.
932 The applicant’s submissions under this section were brief. He submitted as follows:
· MSDA supplied Vioxx to pharmacies which acquired it for re‑supply;
· Vioxx was supplied to group members including the applicant by pharmacies;
· Vioxx was acquired by the applicant for the particular purpose of consumption as a non‑steroidal anti‑inflammatory drug the consumption of which would not lead to a material increase in the risk of suffering life‑threatening conditions …;
· the purpose of acquisition of Vioxx by the applicant (and all group members) was known to MSDA – the very purpose for which Vioxx was developed was to provide an arthritis medication without the potentially serious gastrointestinal side effects of traditional NSAIDs and MSDA’s marketing campaign promoted Vioxx as having a gastrointestinal and overall safety profile which was superior to traditional NSAIDS;
· because Vioxx materially increased the risk of experiencing life‑threatening cardiovascular conditions, Vioxx tablets were not reasonably fit for their purpose of acquisition;
· the applicant suffered his heart attack because Vioxx materially increased the risk of suffering that cardiovascular condition.
933 The respondents’ first submission in response to the applicant’s case under s 74B was one which they made also in relation to s 74D. They submitted that neither section applied to prescription medicines. They submitted that the remedies created by ss 74B and 74D were not “apt to be applied to circumstances [which] are quite remote from the ordinary supply and acquisition of consumer goods”. According to the respondents, the sections imposed norms of conduct which were not “apt to be applied to a case about the inherent properties of a prescription medicine”. That was said to be so because “such goods carry with them inherent risks of side‑effects” and because “their supply is mediated by learned intermediaries, that is, the prescribing doctor and the dispensing pharmacist”. If these general submissions were intended to be anything more than an introduction to the respondents’ case under ss 74B and 74D, they were not developed for that purpose. Indeed, at that general level, the respondents made no submission in addition to those which I have briefly summarised in this paragraph. However, the fact that the goods in question may only be supplied on prescription has the potential to be an important consideration, both under s 74B and under s 74D. I shall return to that consideration in due course.
934 The respondents’ first point under s 74B specifically related to what was said to be the intrinsic inappropriateness of the section to the context of a representative proceeding. The respondents referred to the “individualised nature of the inquiry involved”, relying in this respect on the judgment of Wilcox J in Ryan v Great Lakes Council [1999] FCA 177. That case concerned oysters which had been contaminated by the presence of a foreign substance in the water in which they were grown. In a representative proceeding, the applicant succeeded under both s 74B and s 74D in relation to his personal claims, but Wilcox J held over the claims of the represented parties. As to the application of s 74B in the circumstances, his Honour said (at [368]):
The applicant is entitled to succeed under s 74B as against Barclay Oysters in respect of his personal claim. I cannot make any concluded finding in relation to the applicant's representative claim under s 74B against Barclay Oysters; it is conceivable – although, perhaps, unlikely – that something was said or done, at the time of the supply of oysters to a particular consumer, to make it unreasonable for that consumer to rely on the skill or judgment of Barclay Oysters. The application of s 74B to group members must be left for future determination, if that should prove necessary.
It will be noted that Wilcox J’s reservation as expressed related to the question whether it was unreasonable for a particular consumer to have relied on the skill or judgment of the manufacturer. That is the second aspect of the defence arising under para (b) of s 74B(2). In the present case, MSDA pleaded that aspect of the paragraph in its Defence, but made no submission about it (in relation to the applicant). The particular respect in which Wilcox J held that the claims of the group members in Ryan were heterogeneous does not, therefore, arise on the applicant’s own claim in the present case.
935 Notwithstanding that point of distinction, I accept that the claims of all group members cannot be fully resolved by reference to the case as it has been conducted to date. By the orders made on 30 March 2009 set out in Appendix A to these reasons, the court is presently confined to the resolution of questions which clearly stop a good distance short of dealing with the claims of group members other than the applicant. It is sufficient to say that, for any one group member to succeed under s 74B, he or she must establish what was his or her purpose in acquiring Vioxx and facts sufficient to show, or to give rise to an implication, that that purpose was made known to MSDA. Although, intuitively, the applicant’s own circumstances look as though they might be typical, the claims of the other group members cannot be decided by intuition. Thus I accept that it will be necessary to stand those claims over to a later hearing of the proceeding.
936 The respondents then submitted that, in the context of prescription medicines, there is no “supply” of goods within the terms of s 74B(1)(b). They contended:
Rather, what has occurred is a supply of professional services with the provision of Vioxx as ancillary for these services. That is, there was a trilateral transaction whereby a doctor has prescribed and a pharmacist has dispensed a prescription medicine to a consumer.
There are indeed three parties involved in the transactions out of which a liability presumptively arises under s 74B. However, they are not the parties referred to in the respondents’ submissions: they are MSDA, the pharmacist and the consumer. That a pharmacist supplied Vioxx tablets to the applicant within the meaning of para (b) of s 74B(1) does, with respect to the respondents, appear rather obvious.
937 The respondents relied on E v Australian Red Cross Society (1991) 31 FCR 299, in which the appellant was given infected blood in the course of a transfusion carried out while he was a patient in hospital. Relevantly to the present point, the question was whether the hospital had supplied goods within the meaning of s 71(1) of the Trade Practices Act. With the agreement of Sheppard and Pincus JJ, Lockhart J said (31 FCR 299 at 306):
In my opinion his Honour correctly found that there was no contract with the CSAHS for the supply of blood plasma to the appellant. The essence of the contract between the appellant and the CSAHS was one for services, namely, the provision of hospital, medical and nursing services for the purpose of treating the appellant for his medical problem and restoring him to health. To the extent that goods were provided to him such as food, sleeping tablets, antibiotics, dressings and things of this nature, they were provided as an incident to the contract for the provision of services. There was no contract for the supply of goods. The contract between the CSAHS and the appellant was one for services and is not divisible into a contract for services and for the supply of goods. I leave open, however, the question whether in an appropriate case a contract between a patient and a hospital may be divisible in other circumstances. But the provision on the facts of this case of medicines, drugs and blood cannot be severed into concepts of the provision of services on the one hand and of purchase and sale of goods on the other.
This authority is of no assistance to the respondents in the present case. The transactions with which Lockhart J was concerned related to undifferentiated treatment – which included the supply of blood – given to an inpatient in hospital. A transaction of that character, in my view, is in no sense analogous to that in which a consumer purchases medicines from a pharmacist, albeit that he or she does so on the authority of his or her prescribing doctor. I consider that the Vioxx tablets consumed by the applicant were supplied to him as goods within the meaning of s 74B(1)(b) of the Trade Practices Act.
938 The respondents next submitted that prescription medicines were not “acquired” within the meaning of s 74B(1)(c). Here the presumptive acquisition of goods was that by which the applicant, for example, obtained his Vioxx tablets from a pharmacist. The respondents invoked s 4C(a) of the Trade Practices Act, which provides that “a reference to the acquisition of goods includes a reference to the acquisition of property in, or rights in relation to, goods in pursuance of a supply of the goods”. The respondents submitted that this “definition” (as s 4C(a) was described) was not satisfied because, first, there was no “acquisition of property in or rights in relation to Vioxx” and, secondly, there had been no “supply” of Vioxx as contemplated by the Trade Practices Act. I need say nothing further about the second of these points, as I have already held that the transaction by which the applicant obtained Vioxx tablets from his pharmacist was a “supply” in the relevant sense.
939 The provisions of s 4C(a) of the Trade Practices Act do not affect the ordinary meaning of the word “acquire”. Their only effect is to incorporate into the connotation of that word, where used in the Act, the additional meanings referred to. In the facts of the present case, it is as clear as may be that the applicant acquired Vioxx tablets from his pharmacist, according to the ordinary sense of the word “acquire”. In the circumstances, the applicant has no need to rely (and, indeed, does not seek to rely) on s 4C(a). The respondents’ submissions about that paragraph are, therefore, no answer to the applicant’s case to the extent that it relies upon the provisions of para (c) of s 74B(1).
940 The respondents’ next point was that the purpose for which the applicant alleges that he acquired Vioxx tablets does not reflect his actual purpose at the time, as disclosed by the evidence. As to the latter, the respondents referred to the following passage in the applicant’s witness statement:
I would always take anti‑inflammatory drugs as directed. In consulting with my doctor about taking a drug, I would try to find a balance between a good anti‑inflammatory effect and a low level of gastric discomfort.
They referred also to the following evidence given by the applicant under cross‑examination:
Can I suggest to you that as you over time took anti‑inflammatory drugs you saw yourself as really engaging in a process of taking the drugs because the benefit of them outweighed the identified and known disadvantages to you?---Yes.
Can you tell me this. Did any of the range of doctors who prescribed to you an anti‑inflammatory drug ever tell you, either because they volunteered it or because you asked, what risk, if any, these drugs had with respect to your cardiovascular system?---No.
None at all?---Not that I can remember, no.
If it was said it didn’t cause you to retain it?---I can’t remember it being said.
And the issue of whether there was any cardiovascular risk with respect to your taking of these anti‑inflammatories was therefore never an issue for you to weigh up?---I can’t remember ever having that as a consideration, no.
So you were weighing up pain on the one hand as against no pain and gastrointestinal difficulties on the other?---Basically, yes.
And of course ability to work and do your work without interruption. Is that right?---Yes.
Notwithstanding their reference to these passages, the respondents did not identify the “particular purpose” for which, in their submission, the applicant did acquire Vioxx.
941 The terms of s 74B(1)(c) of the Trade Practices Act allow for a situation in which the acquirer makes known his or her purpose by implication. The implied purpose may be so made known either directly or through the intermediaries referred to in para (c) of subs (1). That such possibilities are allowed under the section introduces scope for the ex‑post formulation of purpose by reference to the needs of the particular litigation in which the remedy is being pursued. According to the respondents, that is exactly what the applicant is attempting to do in the present case. They submit that he has crafted an allegation of his own purpose tendentiously with an astute eye upon the outcome which he seeks in the case. They point out that the identification of purpose in s 74B(1)(c) is necessarily a subjective matter (see Rasell v Cavalier Marketing (Australia) Pty Ltd [1991] 2 Qd R 323, 330), and the applicant’s evidence, set out above, provides no support for the notion that he had in his conscious thoughts the need to obtain a drug “the consumption of which would not lead to a material increase in the risk of suffering life‑threatening conditions”.
942 The authorities show, however, that the putative acquirer does not need to have freedom from the particular defect consciously in his or her mind at the time of acquisition. Thus, for example, Dr Grant was not required to prove that he had in mind purchasing a pair of underpants that would not give him dermatitis: see Grant v Australian Knitting Mills Ltd [1936] AC 85. The fact that he contracted dermatitis from a chemical contained in the material of the garment which he wore justified the conclusion that the garment was not fit for the purpose of being worn as intended (ie against the skin). In the present case, if the applicant’s purpose were no more than to obtain relief from the pain associated with arthritis (and for present purposes it does not matter whether it be osteo or rheumatoid), he need not also demonstrate that he had it in mind that Vioxx would not cause heart attacks. The debate would then move to the question whether, by reason of the association of Vioxx with heart attacks, Vioxx was unfit for the applicant’s purpose. I consider that it is as clear, and as uncontroversial, as may be that the applicant’s purpose in taking Vioxx was to obtain relief from his back pain without (so far as possible) gastrointestinal side‑effects. I am satisfied that his pleading fairly reflected the case which he ran in this regard, and that the respondents were left in no doubt as to the purpose for which he would contend (in his final submissions) that he consumed Vioxx.
943 On the other hand, I do accept the respondents’ submissions that the applicant’s allegation that his purpose was to consume Vioxx as an anti‑inflammatory drug the consumption of which would not lead to a material increase in the risk of suffering life‑threatening conditions is altogether too tendentious an identification of his purpose in taking Vioxx. He gave no evidence that he had such a purpose in that period. It may be said, I suppose, that the purpose of avoiding life‑threatening conditions may be assumed, but this would be, in my view, to make a statement about the applicant’s unstated assumptions, rather than about his purpose. In point of fact, therefore, I reject the applicant’s case that his purpose was to have a drug that did not give rise to a material increase in the risk of suffering life‑threatening conditions.
944 In my view, in a case such as the present where there is no express or clearly implicit expression of purpose that the consumer requires a product that is free of risk (or of increased risk, or materially increased risk, etc), the point at which the absence of risk becomes relevant is where the existence of the presumptively dangerous tendency of the goods in question compromises their fitness for the purpose in fact made known to the manufacturer. For example, a sunscreen lotion that was coincidentally attractive to wasps would be unfit for purpose as a sunscreen lotion. It would be quite artificial to enquire whether the consumer made it implicitly known to the manufacturer that he or she entertained the purpose of using a wasp‑free lotion. On the facts of the present case, the applicant’s point must be that the tendency of Vioxx to contribute to the onset of cardiovascular disease justifies the conclusion that it was not reasonably fit for the purpose of providing relief from arthritic pain.
945 The respondents also submitted that the applicant’s purpose was not “made known” to MSDA, as required by s 74B(1)(c). However, the only submission directly made on this subject was the following:
In these proceedings, there is no evidence that the Applicant’s particular purpose for obtaining Vioxx (let along any Group Member’s) was made known to the First Respondent. For example, Vioxx was at various times approved for the treatment of two indications, the symptomatic treatment of osteoarthritis and rheumatoid arthritis in December 1999 and January 2002 and there is no evidence whatsoever that it was ever made known to MSDA, in respect of any Group Member, for which approved indication Vioxx was prescribed.
This submission, contained in the respondents’ final written submissions, was the subject of neither elaboration nor comment in oral submissions made on their behalf. If the respondents are to be understood as proposing that the applicant’s purpose could not have been made known to MSDA under s 74B(1)(c) unless it knew that he was suffering from osteoarthritis or rheumatoid arthritis, I would have no hesitation in rejecting it. Relevantly to the context of the present case, the particular form of inflammatory disease for which the applicant required Vioxx was quite irrelevant to the kind of unfitness for purpose which he alleges. Vioxx was sold as a product to be consumed with a view to obtaining relief from the pain associated with arthritis. That purpose was inherent and self‑evident. It could not but have been made known implicitly to MSDA that the purpose of every person who was prescribed, and subsequently purchased, Vioxx was that purpose, much as the purpose of a householder who purchases milk is to consume it: Frost v Aylesbury Dairy Company Ltd [1905] 1 KB 608, 612, 614. It is true that (at least after January 2002) it may not have been known to MSDA whether a particular patient was suffering from osteoarthritis or rheumatoid arthritis but, for the reasons I have attempted to explain, that would be neither here nor there (much as the defendant in Frost need not have known the particular purpose to which the contaminated milk was to be put – drinking, cooking etc).
946 At this point it is convenient to return to the circumstance that Vioxx was a prescription‑only medicine upon which the respondents rely. In my view, this does put a particular complexion on the matter of “purpose” under s 74B(1)(c). In a case in which the consumer’s purpose is to be implied by reason of the nature of the goods, the implication must, in my view, take account of the prescription which authorises supply of the goods, and of the fact that it has been written by a medical practitioner. The consumer’s implicit purpose, in such a case, is not merely the achievement of the beneficial effect of the medicine. It is the achievement of that effect in accordance with the terms of the prescription and any advice or instructions that may be assumed to have been given by the doctor, consistently with the prescription. In a clear case, for example, if the doctor had informed the patient that the consumption of a particular medicine carried the real risk of stomach cramp, and if the patient nonetheless decided to take the medicine, his or her implied purpose would be to achieve the conventional beneficial effect of the medicine subject to that risk. It could not then be said that the medicine was not reasonably fit for the purpose as implicitly made known by the consumer.
947 The next point of significance in this respect is that it is the manufacturer to whom the consumer’s purpose must be made known under s 74B(1)(c). In most cases, there will be no direct transaction as between the consumer and the manufacturer, and therefore no communication of the consumer’s actual purpose. Such purpose as is implicit in the nature of the goods will, therefore, be the basis of the consumer’s case. The question will be: considering all relevant circumstances, what communication of purpose as between the consumer and the manufacturer should be implied? In the case of a prescription medicine, those circumstances include those to which I referred in the previous paragraph. So far as the manufacturer’s implied understanding of the consumer’s purpose is concerned, it would, in my opinion, be wrong to omit consideration of any communication made by it to the doctor with respect to the purpose for which the medicine might be fit, or with respect to any adverse consequences of taking the medicine. Again to take a clear case, if the manufacturer had informed the doctor that the medicine carried the real risk of stomach cramp, it could not be implied as between the manufacturer and one of the doctor’s patients (for whom a prescription had been written) that the latter’s purpose was not qualified by reference to that risk.
948 As a matter of principle, it seems that what I have just said should be applicable as much in a case in which the doctor had not in fact advised the patient of the risk that would compromise the medicine’s fitness for purpose as in a case where such advice had been given. That is because we are here concerned with the matter of implication. The question is not what the consumer said to the doctor, but what he or she implicitly “made known” to the manufacturer. It could not, in my view, be held that a manufacturer implicitly received a communication from a consumer save by reference to facts actually or constructively known to the manufacturer at the time. If the manufacturer had advised the doctor of the risk involved in the taking of a particular medicine, that the consumer’s purpose was to derive the beneficial effect of the medicine unaffected by that risk could not, in my view, be implied by the circumstance that the consumer presented the doctor’s prescription to a pharmacist in order to acquire the medicine.
949 The approach which I take to this question is, I think, similar to that taken by Kiefel J in Carey‑Hazell. In that case, a mechanical valve had been surgically inserted into the heart of the patient concerned. It was known that mechanical valves gave rise to the risk of thromboembolisms. Notwithstanding the administration of anti‑coagulants to counter that tendency, the patient did suffer thromboembolic events, and sued the Australian importer of the valve. Kiefel J rejected her case under s 74B. Her Honour said:
212 … The … valve cannot however be regarded as unfit for that purpose because there was a known risk that thromboembolisms might develop and cause injury of the kind the applicant suffered. The applicant has identified only that she fell within the category of persons who develop such a complication. The question whether goods which have a use are reasonably fit for it must be assessed not only by reference to the fact that they failed to accomplish their purpose, but also by reference to what a consumer could reasonably expect from the goods.
213 The evidence here clearly establishes that the risk in question was well known to medical practitioners. The applicant was advised of this risk, as I later discuss. In my view, for the reasons I give later it could not therefore have been reasonable for the applicant to expect that there was no prospect that the valve would cause the development of thrombi. This claim is not made out.
The essence of this passage, in my respectful view, is that the purpose which the consumer implicitly makes known to the manufacturer will necessarily take account of risks that were known to medical practitioners generally.
950 That brings me to the question whether the increased risk of myocardial infarction which I have held to be associated with the consumption of Vioxx was known to medical practitioners generally, or was known to Dr Dickman, either actually or by reference to communications made by MSDA to them or to him. The literal answer to all these questions must be no. MSDA did not know that Vioxx increased the risk of myocardial infarction by a factor of about 2, and it conveyed no information to Dr Dickman, or to the profession generally, about a risk of that order or of anything like that order.
951 The respondents, however, rely on the terms of the Product Information in this context. As to the situation before November 2001 (when the Product Information was amended), there was a note in the “Precautions” section to the effect that, in some trials, myocardial infarctions had been observed to occur (from any cause) in fewer than 0.1% of the patients taking Vioxx. This was not, in my view, the equivalent of a statement that Vioxx should be regarded as itself associated with a risk of myocardial infarction. The respondents did not submit otherwise. Down to November 2001, therefore, there is no way that the purpose implicitly made known to MSDA by the applicant could be regarded as subject to a proviso that a doubling of the risk of heart attack was acceptable.
952 The Product Information as amended in November 2001 provided two further, arguably relevant, pieces of information to medical practitioners who read it: (1) that a trial of Vioxx had shown a lower rate of serious cardiovascular thromboembolic adverse events for patients taking naproxen than for those taking Vioxx; and (2) that, in other trials, spontaneous reports of such events were similar as between Vioxx and non‑selective NSAIDs. The reader was advised to give consideration to those data in the case of an individual patient at risk for such events. Nothing was said here about the risks associated with the consumption of Vioxx as such (ie by reference to a situation in which the notional comparator was placebo). There was nothing to suggest that the consumption of Vioxx, as such, almost doubled the risk of myocardial infarction. Indeed, the earlier reference to which I have referred in the previous paragraph remained in the Product Information. I do not consider that the amended Product Information provided any basis for saying that the purpose of acquiring Vioxx implicitly made known by the applicant to MSDA was subject to a qualification that a doubling of that risk was acceptable.
953 Thus, although I reject the applicant’s point that the purpose which he implicitly made known to MSDA was the consumption of a drug which did not lead to a material increase in the risk of suffering life‑threatening conditions, I also reject the respondents’ point that his purpose, as so communicated, was qualified to an extent that made acceptable anything like a twofold increase in the risk of suffering a myocardial infarction.
954 The respondents’ next point under s 74B called up the defence arising under para (b) of subs (2). They submitted that the applicant did not rely on MSDA’s skill or judgment. Rather, he was said to have relied wholly on the skill or judgment of his treating practitioner, Dr Dickman. I note in this context that MSDA did not advance a defence under the second limb of subs (2)(b) (ie that it was unreasonable of the applicant to have relied on its skill or judgment).
955 Undoubtedly the applicant relied on the skill or judgment of Dr Dickman. But that is not the end of the matter. Neither is it the question posited by para (b) of subs (2). A consumer will often rely on the skill or judgment of a specialised retailer in deciding, for example, which brand or model of particular goods to purchase, but that circumstance is not inconsistent with the consumer at the same time implicitly relying on the skill or judgment of the manufacturer.
956 The requirement of s 74B(2)(b) is not that the consumer did not rely on the skill or judgment of the manufacturer, but that the circumstances show that he or she did not so rely. This proposes a highly objective approach to the matter of reliance. It is almost as though the legislature contemplated that a consumer might, ex‑post, give very self‑serving evidence about the matter. However, the paragraph operates by way of a defence. The applicant does not have to establish that he relied on MSDA’s skill or judgment: see Vines v Djordjevitch (1955) 91 CLR 512, 519‑520; Chugg v Pacific Dunlop Ltd (1990) 170 CLR 249. At the core of the defence is the consumer’s own state of mind at the relevant time. It would, in my view, be an unusual case in which a court would uphold the defence by reference to a matrix of circumstances which had not been put to the consumer in cross‑examination, or where it had not been put to the consumer that he or she did not in fact rely on the manufacturer’s skill or judgment.
957 In the present case, the applicant was not, in cross‑examination, confronted with the circumstances said to justify the conclusion that he did not rely on MSDA’s skill or judgment. Neither was it put to him that he did not so rely. Indeed, it is hard to see how the defence now sought to be advanced by MSDA is consistent with the flow of the following cross‑examination of the applicant:
It’s right to say, isn’t it, that as you understood it in May 2001 that Vioxx was a prescription medication that would, just like your anti‑inflammatories, relieve the pain from which you were suffering?---Correct.
But it would have an added benefit of addressing your gastrointestinal difficulties?---Correct.
You understood that it didn’t have at that time – at that time your understanding was that such side effects as they had, whatever they were, would be no different to the anti‑inflammatoriesthat you had been taking?---Nodifferentotherthannogastric---
Correct but the disadvantageous side effects were no different than the anti‑inflammatory medications you had been taking and would have a particular benefit?---Yes.
As I have said, the applicant left no doubt but that he relied on the skill or judgment of Dr Dickman. If it were the respondents’ case that the applicant did not, therefore, also rely on the skill or judgment of MSDA, I consider that such a proposition ought to have been put to the applicant directly; or at least the “circumstances” that would later be relied on to show that he did not so rely should have been put to him. Neither was done.
958 For the above reasons, I reject MSDA’s reliance on s 74B(2)(b) of the Trade Practices Act.
959 For the sake of completeness, I observe that the respondents approached the question arising under s 74B(2)(b) as though the “consumer” there referred to was the applicant. No submission was advanced in the alternative against the prospect that I might find that the Vioxx tablets which the applicant consumed in the period immediately preceding his heart attack in December 2003 were obtained on a prescription written for his wife and that she, therefore, was, or may have been, the relevant “consumer”. If so, to the extent that the applicant suffered loss or damage as a result of consuming the tablets, he would be the “other person” referred to in subs (1). The respondents did not deal with the question whether Mrs Peterson relied on the skill or judgment of MSDA.
960 Finally under s 74B, the respondents took issue with the applicant’s submission that Vioxx was not reasonably fit for the purpose which he implicitly communicated to MSDA. Specifically, they proposed that, since prescription medicines notoriously had the potential to involve side-effects, Vioxx should not be regarded as unfit for purpose merely by reason of such a circumstance.
961 The applicant’s case is that Vioxx was not fit for purpose because it gave rise to an increase in the risk of encountering potentially life‑threatening conditions. Objectively, I have found that Vioxx did give rise to about a doubling of the risk of suffering myocardial infarction, across a population. At the simplest level, there could be little dispute about the proposition that a drug for the relief of arthritic pain is not fit for that purpose if the consumption of it will cause the patient concerned to have a heart attack. That is not the present case, of course, but it serves as a base line for a consideration of the position of a drug which gives rise to a risk, or which increases a pre‑existing risk, of heart attack.
962 Section 74B is concerned with the objective fitness of goods for purpose, whether or not the circumstance presumptively preventing them from qualifying for that description was known at the time of supply. In Medtel Pty Ltd v Courtney (2003) 130 FCR 182, 206, [70], Branson J said (with assent of Jacobson J):
Section 74D, … calls for the quality, or fitness for purpose, of the goods to be measured against what it was reasonable to expect in that regard at the time of the supply of the goods to the consumer. That measurement must be undertaken, in my view, in the light of information concerning the goods available at the time of trial. However, the issue remains whether the goods were as fit for the relevant purpose as it was reasonable to expect at the time of their supply to the consumer.
Mutatis mutandis, the same approach is, in my view, required under s 74B. In the context of the present case, this means that the fitness of Vioxx for purpose must be assessed against the fact that it gave rise to an increase in the risk of myocardial infarction by a factor of about 2: see para 570 above.
963 I did not understand the respondents to contend that goods could not be regarded as unfit for purpose where all that was known was that they involved the risk of an adverse outcome (or an increase in a pre‑existing risk of such an outcome). Any such contention, in my view, could not stand alongside the judgment of the Full Court in Medtel, where a pacemaker which had not failed, and which was regarded by the primary judge as unlikely to fail, was held to be unfit for purpose because it had been made by a production process which was associated with an increased risk of failure. The respondents make the point, which I accept so far as it goes, that Medtel concerned a faulty product (or at least a potentially faulty product), whereas Vioxx tablets in the present case contained no manufacturing or like fault. This is, however, a distinction without a difference with respect to the point now under consideration. What Medtel demonstrates, in my view, is that the fact that goods give rise to a risk of an outcome of a seriously adverse nature is sufficient, other circumstances permitting, to justify the conclusion that the goods are not fit for purpose.
964 It seems clear that Vioxx was efficacious in the relief of arthritic pain, and would remain so notwithstanding that there might be substance in the applicant’s allegation of increased risk. If the notion of purpose is viewed through a narrow prism, there is a sense in which it might be said that Vioxx was fitted for purpose because it provided that relief, whatever might have been the risks associated with its consumption. That is not, however, in my view, an appropriate way to view the concepts arising under s 74B(1)(d). In each case, the basic inquiry is whether the reasonable person would regard the fitness for purpose of Vioxx as compromised by its tendency to produce the unwanted secondary adverse effect. Or, put another way, would the reasonable person reject Vioxx as a suitable drug for the relief of pain if he or she were informed that consumption of it had been found, across a population of persons not shown to be obviously atypical, to double the risk of heart attack? Subject to the other matters with which I deal in these reasons, an inquiry such as this could yield only one answer: an affirmative one.
965 As I have indicated, the respondents submitted that a medicine is not unfit for purpose merely because it carries the risk of side-effects. It was said to be notorious (and the applicant himself accepted) that prescription drugs often carry such risks, yet they are regularly used to beneficial effect without any suggestion they are unfit for purpose or the like. According to the respondents, if Vioxx did carry a cardiovascular risk, that was in the nature of a side‑effect, and it did not mean that the drug was not reasonably fit for the purpose for which it was both prescribed and acquired. The respondents relied particularly on the judgment of Kiefel J in Carey-Hazell. Here I refer to the passages from her Honour’s reasons set out in para 949 above. I do not think that her Honour’s words stand as any authority for the sweeping proposition that the presence of adverse side-effects associated with an otherwise efficacious drug could never be such as to render the drug less than fit for the purpose within the meaning of s 74B(1)(d). It was an essential aspect of her Honour’s reasoning that the risk which came home to the patient in question was the subject of broad knowledge within the medical profession. In the present case, the increase in risk which is alleged to have been associated with the consumption of Vioxx was not generally known.
966 That does not, however, dispose of the respondents’ point that Vioxx should not be regarded as unfit for purpose merely because it had, or potentially had, a side‑effect. In my view, the point is not susceptible of a categorical resolution across all possible cases by reference to the characterisation of the unwanted tendency associated with Vioxx as a “side‑effect”. The question will always be whether that tendency made Vioxx less than reasonably fit for purpose. As stated earlier, the applicant alleged that Vioxx was not reasonably fit for purpose because consumption of it led to an increase in the risk of suffering life-threatening conditions. I consider that it would be no answer to a case presumptively established under s 74B to say that the risk, or the increase in risk, was proper to be regarded as a side-effect. I think it quite improbable that any reasonable person, at the time of supply, would regard as satisfactory a response from the hypothetical pharmacist to the effect, “Don’t worry, it’s only a side‑effect such as most medicines have”.
967 Related to the reasonableness of an expectation that prescription drugs often carry side-effects is, of course, the extent of information or warnings about the side-effect in question which are brought to the attention of the consumer or of his or her doctor. While I accept that the reasonable person in the street would be unsurprised to learn that a particular medicine − any medicine − has, or has the potential for, side-effects, I likewise consider that he or she would assume that such information or warnings would be so provided. The respondents did not tie this part of their submissions, however, to the provision by MSDA of any such information or warnings. They did make a submission about the present relevance of the Vioxx Product Information, and I have dealt with that aspect above.
968 For the above reasons, I am satisfied that, because it approximately doubled the risk of heart attack, Vioxx was not reasonably fit for the purpose implicitly made known to MSDA by the applicant. I express this conclusion without regard to the base-line risk of heart attack which the applicant had at the relevant times. Whatever that risk, in my view a doubling of it would disqualify Vioxx from reasonably meeting the applicant’s implicit purpose. It follows that I shall uphold the applicant’s case under s 74B of the Trade Practices Act, and turn presently to a consideration of compensation as is his entitlement under subs (1) of that provision.
Section 74D
969 Section 74D of the Trade Practices Act provides as follows:
(1) Where:
(a) a corporation, in trade or commerce, supplies goods manufactured by the corporation to another person who acquires the goods for re‑supply;
(b) a person (whether or not the person who acquired the goods from the corporation) supplies the goods (otherwise than by way of sale by auction) to a consumer;
(c) the goods are not of merchantable quality; and
(d) the consumer or a person who acquires the goods from, or derives title to the goods through or under, the consumer suffers loss or damage by reason that the goods are not of merchantable quality;
the corporation is liable to compensate the consumer or that other person for the loss or damage and the consumer or that other person may recover the amount of the compensation by action against the corporation in a court of competent jurisdiction.
(2) Subsection (1) does not apply:
(a) if the goods are not of merchantable quality by reason of:
(i) an act or default of any person (not being the corporation or a servant or agent of the corporation); or
(ii) a cause independent of human control;
occurring after the goods have left the control of the corporation;
(b) as regards defects specifically drawn to the consumer’s attention before the making of the contract for the supply of the goods to the consumer; or
(c) if the consumer examines the goods before that contract is made, as regards defects that the examination ought to reveal.
(3) Goods of any kind are of merchantable quality within the meaning of this section if they are as fit for the purpose or purposes for which goods of that kind are commonly bought as it is reasonable to expect having regard to:
(a) any description applied to the goods by the corporation;
(b) the price received by the corporation for the goods (if relevant); and
(c) all the other relevant circumstances.
970 The applicant submitted that the meaning of the term “merchantable quality” was given by subs (3) of s 74D and that, in determining what it was objectively reasonable for the consumer to expect, the court had to take into account all relevant information available at the time of the trial: the court was not limited to a consideration of the information available at the time of supply to the consumer. Nor, according to the applicant, was it relevant whether the manufacturer had any means of knowing, at the time of supply, that the relevant goods were not of merchantable quality. The applicant’s submissions continued:
· MSDA supplied Vioxx tablets to pharmacies;
· pharmacies supplied Vioxx tablets to the applicant and other group members;
· it was objectively reasonable to expect that Vioxx would be fit for the purpose of consumption as a drug for the treatment of arthritis which did not materially increase the risk of potentially life‑threatening conditions – after all, the purpose for which Vioxx was developed was to provide an arthritis medication without the potentially serious gastrointestinal side effects of traditional NSAIDs and MSDA’s marketing campaign promoted Vioxx as having a gastrointestinal and overall safety profile which was superior to traditional NSAIDS;
· the Vioxx tablets were not of merchantable quality in that the consumption of Vioxx materially increased the risk of suffering life‑threatening cardiovascular conditions.
971 Because of the terms of s 74D(3), many of the questions arising under the section correspond with the like questions arising under s 74B with which I have dealt above. The differentiating circumstance as between the two sections lies in paras (c) and (d) of s 74B(1) and para (c) of s 74D(1). Under the former, the goods must have been acquired for a purpose that was made known to the manufacturer. If the goods are not reasonably fit for that purpose, the manufacturer is liable. Under the latter, the purpose for which the goods were acquired is irrelevant: if they are not as fit for the purpose or purposes for which goods of the kind in question are commonly bought as it is reasonable to expect, having regard to the matters set out in s 74D(3), the manufacturer is liable. However, the inherent concept of fitness for purpose is the same in each section. Further, I consider that the objective test of reasonable fitness under s 74B(1)(d) is relevantly indistinguishable from the like test of what degree of fitness is reasonable to expect under s 74D(3). In the present case, no party submitted otherwise.
972 As they said about s 74B, the respondents likewise submitted that the claims of parties in a representative proceeding under s 74D could not be resolved without an individualised consideration of the position of each group member. At least in a case in which none of the defences referred to in subs (2) is raised, at first blush it looks as though the position arising under s 74D would differ from that arising under s 74B, because the condition of merchantable quality of the goods in question is not related to the purpose of the consumer in question. However, in Ryan, Wilcox J thought otherwise. His Honour said ([1999] FCA 177 at [375]):
The s 74D claim should be determined in the same way as that arising under s 74B: the applicant is entitled to succeed on his own behalf against Barclay Oysters, although not Barclay Distributors. His representative claim against Barclay Oysters should be reserved.
Although his Honour did not state the reasoning behind the second sentence in this extract, I accept that it is theoretically possible that facts which might be brought forward under paras (a) and (c) of s 74D(3) for, or against, other group members would not be the same as those to which the court has been exposed on the case concerning the applicant himself. Thus, like Wilcox J, I shall stand over the s 74D claims of the other group members.
973 The respondents made the same point about there being no “supply” of goods within the meaning of s 74D(1)(b) in the circumstances of prescription medicines as they had under s 74B(1)(b). I would decide the point the same way as I did in paras 936‑937 above.
974 Turning to the “acquisition” point in its application to s 74D, the respondents submitted that s 74D(1)(d), like s 74B(1)(c), required that “the goods [be] acquired by the consumer”. (The respondents actually made this submission about an entirely irrelevant provision of the Trade Practices Act, s 75B(1)(d), but I take that to be a typographical error, and shall read it as a reference to s 74D(1)(d)). However, s 74D(1)(d) imposes no such requirement. To the extent that it deals with an acquisition of goods, the paragraph is concerned with a person – not the consumer – who acquires the goods from the consumer. In addition to the reasons given above for rejecting the “acquisition” point in relation to s 74B(1)(c), I would also hold that the point, in relation to s 74D(1)(d), is misconceived. For the sake of completeness, I would add that no submission was made by the respondents that, should I find that the applicant consumed tablets obtained under his wife’s prescription in November and early December 2003, he did not “acquire” those tablets from her within the meaning of s 74D(1)(d).
975 I turn next to the central question arising under s 74D, namely whether the Vioxx tablets taken by the applicant were as fit for the purpose (or purposes) for which goods of that kind were commonly bought as it was reasonable to expect. For reasons given above in the context of s 74B, I consider that the Vioxx tablets taken by the applicant were not as fit for the purpose for which goods of that kind were commonly bought as it was reasonable to expect because they increased the risk of heart attack by a factor of about 2. It was, in my view, reasonable to expect that a medication for the relief of arthritic pain would not carry that risk. As with s 74B, I do not think it is any answer to this point to say that the risk was in the nature of a “side-effect”.
976 I next mention three aspects of the present question upon which I was not addressed by the parties, but which I have not overlooked. The first relates to the presently material connotation of the expression “goods of that kind” in subs (3). It does seem as though the provision requires one to identify a class to which the goods in question belong, and to consider the purpose for which goods of that class are commonly bought. There may be a question, for example, whether the “kind” of goods that is relevant in the present case is prescription medicines, or prescription medicines for the relief of pain, or prescription medicines for the relief of arthritic pain. There may also be a question whether the “kind” of goods which should be regarded as relevant in the case of Vioxx was NSAIDs generally or coxibs in particular. If the latter, would Vioxx have been less fitted for purpose than was reasonable to expect if, in a particular case, consumption of it gave rise to gastrointestinal complications? These questions, interesting though they are, do not arise in the present case as, whether the connotation of “kind of goods” be narrow or broad, the complaint made by the applicant is that Vioxx was less fitted for purpose than it was reasonable to expect by reason of a circumstance that would likewise disqualify any medicine for the symptomatic relief of arthritic pain. It is sufficient, in other words, to say that Vioxx was a member of a class of medicines which were commonly bought for that purpose.
977 The second point is whether any description “applied to the goods” by MSDA bears upon the resolution of the question whether Vioxx was as fit for purpose as it was reasonable to expect. As I have observed elsewhere, no party tendered the packet in which Vioxx was sold. Nor was a copy of the Consumer Medicine Information put in evidence, although different drafts of the text for that document for which the approval of the TGA was sought were. There is nothing in those drafts, or in the evidence otherwise, that would tend to indicate a conclusion under s 74D(3) different from that which would otherwise be drawn. To the extent that it might be considered that the Product Information was “applied to the goods” within the meaning of para (a) of that subsection, I refer to my reasons in paras 951‑952 above. In the result, if the court would otherwise reach the conclusion that Vioxx was not as fit for the purpose for which drugs for the symptomatic relief of arthritic pain were commonly bought as was reasonable to expect, I do not consider that such a conclusion is to be qualified by reference to any description applied to the goods.
978 The third matter which I mention for completeness merely involves noting that the respondents did not ask me to find that Vioxx tablets were as fit for the purpose for which goods of their kind were commonly bought because NSAIDs generally carried a risk of myocardial infarction; nor because all coxibs carried such a risk to the same or a similar extent as did Vioxx; nor because the risk was not reasonably discernable by MSDA by reference to the state of scientific understanding at the relevant times.
979 The respondents made five further (very brief) submissions about the applicability of s 74D to the circumstances of the applicant. First, they submitted that, under s 74D(3), goods do not have to be absolutely fit for their general purpose: it is sufficient if they are, objectively, as fit for that purpose as it is reasonable to expect. I accept that submission. So, as I understand it, would the applicant. In my disposition of the applicant’s case under s 74D, I have approached the matter as here proposed by the respondents.
980 Secondly, the respondents submitted that, as a matter of law, prescription medicines made available in accordance with the terms of an approval from the TGA are of merchantable quality for the purposes of s 74D. This point was not developed, elaborated upon, or explained either in writing or orally. The court, in my view, is under no obligation to attempt to divine what might be the content of, or the jurisprudential justification for, a point so inadequately presented as this one. I reject it.
981 Thirdly, the respondents submitted that the feature of Vioxx alleged to render it unmerchantable was not an error in formulation or manufacture, but was inherent in the proper formulation of the drug for its intended purposes. I have effectively dealt with this point in my consideration of the judgment of Branson J in Medtel, in para 963 above. To elaborate upon what I said there, I do not accept that the only basis upon which goods may be less than as fit for purpose as it is reasonable to expect is because of a manufacturing or like fault, or some departure from specification. Prescription drugs, where the object is to modify the body’s biochemistry, provide a conspicuous example of goods which may be faultless so far as specification and production quality are concerned, but which may be unfit for purpose because of some unintended, but inherent, effect which they have. In my view, there is nothing in this point which would stand in the way of the applicant succeeding under s 74D.
982 Fourthly, the respondents submitted that the fact that a product had been included in a hazard alert or product recall notice was not alone sufficient to render the product unmerchantable. I accept this point. Nothing in these reasons relies upon the withdrawal of Vioxx from the market in September 2004 as a basis for upholding the applicant’s case under s 74D.
983 Fifthly, there was one further point which the respondents sought to raise in opposition to the applicant’s case under s 74D. It involved reliance on paras (b) and (c) of subs (2). If good, the point arises by way of a defence, and was not pleaded. I do not consider that it is open to the respondents to rely on those paragraphs. As it happens, neither in the brief submissions relevantly made by the respondents nor in any of the evidence is there support for either of the circumstances postulated by paras (b) and (c). I would find as a fact that the tendency of Vioxx to give rise to an increased risk of heart attack was not specifically drawn to the applicant’s attention before he purchased (or his wife purchased) any of the Vioxx tablets the consumption of which gave rise to that increase in risk; and that no examination of the tablets by the applicant would have, or could have, revealed that tendency.
984 Save to the extent expressly mentioned above, I would decide the case under s 74D consistently with my decision under s 74B. I uphold the applicant’s case, and shall consider his entitlement to compensation under s 74D(1).
Compensation
985 Under s 74B(1)(e) and s 74D(1)(d) of the Trade Practices Act, the applicant’s entitlement to compensation depends upon the following circumstances:
(1) that he was the consumer referred to in the subsection or a person who acquired the goods from the consumer; and
(2) that he suffered loss or damage by reason that the goods were not reasonably fit for purpose, or were not of merchantable quality.
The “goods” referred to in each of these circumstances are the same goods. That is to say, the loss or damage must have the necessary connection with the goods which were supplied to the applicant as consumer or which he acquired from the consumer.
986 These points, which may appear obvious, are important in the context of the present case because I have held that the Vioxx tablets taken by the applicant did contribute to his heart attack, but that the tablets he took between 3 November and 2 December 2003 were most probably obtained under his wife’s prescription. That means, of course, that, in relation to those tablets, Mrs Peterson was the consumer under ss 74B and 74D, and the applicant was the person who acquired the goods from her. That would still give the applicant a cause of action, and probably nothing turns on the distinction. I mention the point here for the sake of completeness, and note that the respondents made nothing of it in their submissions. Although they submitted that I should find that the applicant took no Vioxx at all between 8 October and 2 December 2003, they made no submission in the alternative to the effect that, if I should find that he took his wife’s tablets after 3 November, those tablets were not properly to be regarded as the “goods” apropos the applicant’s own claim under ss 74B and 74D.
987 Neither did the respondents submit that, if I should find that the Vioxx tablets which the applicant took were not reasonably fit for purpose and/or not of merchantable quality, and that those tablets did contribute to the applicant’s heart attack, the loss or damage presumptively associated with and arising from the heart attack should not be regarded as having occurred by reason of that unfitness for purpose or lack of merchantable quality. They conducted their case on the footing that, if these elements were established by the applicant, the debate could then move to questions of the quantification of the compensation to which the applicant was entitled under ss 74B and 74D. The applicant is, of course, to be compensated once only, notwithstanding that I have held that there is an entitlement arising under each section. That entitlement is the same in each case, and is the matter to which I now turn.
988 In assessing damages for pain, suffering, inconvenience and loss of amenity of life, the following are, in my view, the significant factors. First, the applicant’s heart attack was directly associated with quite severe pain. This occurred over a period of about two hours in the early hours of 8 December 2003. I also infer that this was a frightening experience for him. Secondly, the applicant’s condition required him to be hospitalised, to undergo surgery, to endure a period of recovery and to participate in rehabilitation. Thirdly, the permanent damage to the applicant’s heart muscle was substantial, and has limited him in the range of occupational and recreational activities which he can undertake. He more readily tires from levels of physical activity that would previously have been of little concern to him. His “whole of person impairment” is of the order of 25-30%. Fourthly, he is obliged to adhere to a regime of medication and to implement what Prof Harper described as “lifestyle modification” to ward off the prospect of further heart disease. And fifthly, the damage to his heart has left him in a less robust condition to resist the impact of a future heart attack, should he have one.
989 As against these factors, the surgery which the applicant underwent was, it seems, as routine as could be expected given the serious condition from which he suffered, the outcome was successful and his recovery was uncomplicated. The relief of his stenosis was complete. There is no evidence that the heart attack (as distinct, possibly, from factors such as anxiety) has left the applicant with any periods of pain or discomfort. Subject to the lifestyle modification to which I have referred, and to compliance with the regime of medicines which has been laid out for him, the applicant seems able to lead a normal and comfortable life. To the extent that his ability to work is compromised, I shall consider his past and future loss of earnings capacity below. At what might be regarded as the emotional level, however, the applicant did not complain that an inability to work harder and longer was a source of distress or frustration for him. I note that he continues to operate (with no assistance other than that of his wife) his “Lockout” business, and did not suggest that his capacity to do so had been impaired by his heart attack.
990 The applicant now seems to be at less risk of a further heart attack than he was at risk of the heart attack which he had. Looking at the applicant’s risk factors as at 2001, Prof Harper expressed the view that there was about a 25% risk that the applicant would have a heart attack over the ensuing five years. By December 2003, because of the improvements which the applicant had achieved in his lipid profile (but not taking into account the effect of the consumption of Vioxx), the risk had reduced slightly, to be somewhere in the region 2025%. At present (ie, I assume, as at his consultation with the applicant in October 2008) Prof Harper would estimate the risk of heart attack over the next five years as in the region of 5-10%. No doubt because of this evidence, it was not submitted on behalf of the applicant that the events of 8 December 2003 had made it likely that he would have a further heart attack sooner than would otherwise have been the case.
991 On the other hand, counsel for the applicant did submit that, if the applicant were to have a further heart attack, the one which he had in December 2003 would increase the likelihood of the next one being fatal. That submission is based on the unchallenged evidence of Prof Harper, and I accept it. It does not follow, however, that, overall, the applicant’s expectation of life is less now than it would have been in the absence of the heart attack which he had. For the reasons given in the previous paragraph, and doubtless because of the changes which that heart attack has obliged the applicant to make, I could not find on the probabilities that his expectation of life is less than it presently would have been had he never taken Vioxx.
992 It was submitted on behalf of the applicant that an appropriate award for the kind of general damages which I am now discussing would be in the range $175,000−$250,000. In my view, an award of such an order would go beyond anything that could be regarded as reasonable compensation for the general non-economic loss and damage which the applicant has endured and will endure. The respondents submitted that the figure should be in the range $50,000−$75,000, including interest. This range is, in my view, somewhat more realistic than that proposed on behalf of the applicant, but I should make it clear that I do not propose to enter upon the question of interest in these reasons. I shall receive submissions from the parties in that regard in due course.
993 The applicant sought to rely on s 28HA of the Wrongs Act 1958 (Vic), which provides:
(1) In determining damages for non-economic loss, a court may refer to earlier
decisions of that or other courts for the purpose of establishing the
appropriate award in the proceedings.
(2) For that purpose, the parties to the proceedings or their counsel may
bring the court's attention to awards for damages for non-economic loss in
those earlier proceedings.
(3) This section does not alter the rules for the determination of other
damages.
Despite what I presume is a wealth of precedent in Australia (whether or not relating to cardiovascular disease as such), however, the only “decision” to which the applicant referred me in this regard was Bowling v Pfizer, Inc 143 FRD 141 (1992), a judgment by the United States District court, SD Ohio, Western Division, approving a scheme for the settlement of a class action involving faulty artificial heart valves. The court itself made no decision as to the appropriate level of damages for the injury suffered by any member of the class. The actual entitlement of individual members was, apparently, to be left to an “expert panel”, and the applicant here sought to refer to details of the panel’s determination. But there was no evidence of it. I also note that the damages claimed by the plaintiffs included what the court described as “punitive damages”. Whatever role s 28HA might have played in the present proceeding, the applicant’s reference to Bowling was of no assistance to me.
994 In my view, an appropriate sum by way of compensation for the pain, suffering, inconvenience and loss of amenity of life endured, and to be endured, by the applicant is $85,000.
995 Turning to economic loss, in order to provide an estimate of the loss which the applicant suffered between the date of his heart attack and the trial of the proceeding, he called Mark Thompson, a chartered accountant and a director of Vincent’s Chartered Accountants of Brisbane. Mr Thompson estimated that the applicant’s loss to 30 June 2008 was $134,765. He made that estimate by reference to the following table:
|
|
Jan to June 2004 |
2005 |
2006 |
2007 |
2008 |
|
|
Notional Gross Consulting Fee Income |
50,000 |
125,000 |
150,000 |
175,000 |
195,000 |
|
|
Less: Actual Gross Consulting Fee Income |
28,793 |
48,350 |
88,005 |
100,892 |
156,061 |
|
|
|
|
|
|
|
|
|
|
Loss of Gross Consulting Fee Income |
21,207 |
76,650 |
61,995 |
74,108 |
38,939 |
|
|
|
|
|
|
|
|
|
|
Less:Allowance for Direct expenses Saved (at 17.5%) |
(3,711) |
(13,414) |
(10,849) |
(12,969) |
(6,814) |
|
|
|
|
|
|
|
|
|
|
NET LOSS (before tax) |
17,496 |
63,236 |
51,146 |
61,139 |
32,125 |
|
|
Less: Income Tax applicable |
(8,485) |
(25,674) |
(15,906) |
(25,373) |
(14,938) |
|
|
|
|
|
|
|
|
|
|
NET LOSS (after tax) |
9,010 |
37,562 |
35,240 |
35,766 |
17,187 |
|
|
|
|
|
|
|
|
|
|
TOTAL LOSS TO 30 JUNE 2008 |
|
|
|
|
134,765 |
|
996 The respondents took issue with the first line of Mr Thompson’s table, that relating to “Notional Gross Consulting Fee Income”. That was the level of income which Mr Thompson estimated would have been derived by Mr Peterson from his consulting activities if he had not had a heart attack. Mr Thompson derived these figures as follows. He accepted an estimate given to him by Mr Peterson of the extent to which his consulting activities were curtailed by his having had a heart attack. That estimate was 20%. Mr Thompson then took the applicant’s actual gross consulting fee income for the year ended 30 June 2008 ($156, 061), and treated that as 80% of the income which he would have earned in the absence of his heart attack. That income, therefore, was $195,000. Mr Thompson then assumed that the full-year equivalent for the applicant’s consulting fee income in the year ended 30 June 2004 would have been $100,000. He then, in effect, drew a straight line between that figure and $195,000 to derive the figures set out in the first line of his table above.
997 Under cross-examination, Mr Thompson accepted that he had no documents which would support his assumption that $100,000 was the appropriate starting point for the year ended 30 June 2004. Further, it seems that there was no particular basis for his assumption that the applicant’s consulting fee income would increase at a constant rate between that year and the year ended 30 June 2008. As it happened, by correspondence dated 18 February 2008, Mr Thompson addressed a series of questions to Mr Gibb, one of which was:
How has your business activity increased over the period after you ceased with BHP? This response if possible will provide either dollar value turnover’s [sic] for 2004, 2005, 2006, 2007 and year to date or alternatively % changes over those years.
On 19 February 2008, Mr Gibb replied that his company’s growth had been 36%, 8% and 40% in the financial years to 30 June 2005, 2006 and 2007 respectively. In his oral evidence, Mr Gibb said that the growth had been between 25% and 30% in the year to 30 June 2008 (his best estimate was 27.5%).
998 The respondents submitted that Mr Thompson could have done better than to make what they said was an artificial assumption as to the growth of the consulting fee income which the applicant notionally would have derived between 2004 and 2008. They submitted that the growth figures provided for SWS by Mr Gibb represented the actual experience of a consulting business in the real world, and would have been a more realistic basis for the task of estimation on which Mr Thompson was engaged. What the respondents proposed was that one should commence with the known figure of $195,000 in the year to 30 June 2008, and work back to the year ended 30 June 2004 by the application of Mr Gibb’s percentages. If this were done, instead of the sums appearing in the first line of Mr Thompson’s table as set out above, the income figures would be $37,286, $101,419, $109,533, and $153,346 for the years 2004, 2005, 2006 and 2007 respectively. The total figure to 30 June 2008 would become $84,354.
999 Neither in his counsel’s re-examination of Mr Thompson, nor in his submissions in reply, did the applicant join issue with the respondents with respect to their recalculation of the first line of Mr Thompson’s table. Indeed, in their final submissions, counsel for the applicant referred to Mr Thompson’s original calculations as though there had been no challenge to the basis upon which they were made. I am persuaded that the recalculation proposed by the respondents provides a better estimate of the way in which the applicant’s consulting fee income would have grown in the absence of his heart attack than that originally provided by Mr Thompson. I accept that the percentage figures provided by Mr Gibb represent a steeper growth profile for this kind of work, and require a conclusion that the applicant’s notional annualised consulting fee income for the year ended 30 June 2004 was of the order $75,000, not $100,000. In the circumstances, I accept the respondents’ figure of $84,354 as an estimate of the consulting fee income lost by the applicant down to 30 June 2008.
1000 The respondents made a series of further points by reference to which they proposed that Mr Thompson’s figure for past economic loss (and even the figure of $84,354, as I understand it) should be qualified. The first related to Mr Thompson’s analysis (from the invoices provided by the applicant) of the days of consulting, and the total days of work, undertaken by the applicant in the period to 30 June 2008. In this respect, Mr Thompson provided the following table:
|
Year ended /period |
No. of days consulting |
No. of days travelling |
Total Days |
|
November 2003 to June 2004 |
30.5 |
0.5 |
31.0 |
|
30 June 2005 |
60.75 |
1.0 |
61.75 |
|
30 June 2006 |
55.13 |
9.00 |
64.13 |
|
30 June 2007 |
62.0 |
6.0 |
68.00 |
|
30 June 2008 |
75.0 |
18.0 |
93.00 |
Under cross examination, Mr Thompson accepted that the figures in the column headed “No. of days travelling” related only to the travelling days for which the applicant had rendered an invoice. An examination of the destinations at which the applicant had provided consulting services, however, showed that there were, at least in some cases, days occupied travelling for which the applicant had not charged. These had not been taken into account by Mr Thompson, and, if they were, the figures in the column headed “Total Days” would be higher than those presently appearing. Mr Thompson accepted this criticism of his methodology, but, in their final submissions, the respondents did not make it clear how, if at all, Mr Thompson’s estimate of past economic loss should be adjusted as a result of this circumstance. They submitted that what they described as “unaccounted travel” was “relevant to the levels of consultancy work which the applicant claims he would have been able to undertake” in the absence of his heart attack. However, notwithstanding having cross-examined Mr Thompson by reference to records from which (they submitted) quite reliable inferences could be made as to the additional days of travelling undertaken by the applicant, in their final submissions the respondents provided no overall analysis as to the extent of this problem over the period with which Mr Thompson’s calculations were concerned. All the court knows is that there was, in effect, a day here and a day there in which the applicant had been travelling, but for which he had not rendered an invoice. The respondents contented themselves with submitting that “an appropriate adjustment needs to be made to effect this understatement of the number of days for which Mr Peterson actually was engaged as a consultant”. That was, in my view, an unsatisfactory place to leave the matter, at least if the respondents expected the court to reject some aspect of Mr Thompson’s calculations on account of it. I do not propose to do so.
1001 The respondents’ next point was that the steadily increasing success of the applicant’s “Lockout” business would necessarily have required his personal attention, and thereby have reduced the number of days for which he would have been available to consult. It was the applicant’s case that the “Lockout” business, even as it became more successful, could be managed (with his wife’s assistance) without compromising his availability to consult. It was implicit in that case that, in the absence of his heart attack, the applicant would have undertaken consulting work 20% more than he did, while still deriving the same level of income from his other business. This point requires a broad understanding of the applicant’s “Lockout” business and of what direct attention it required from him.
1002 I described the “Lockout” system at para 694 above. The product actually sold by the applicant (or, more accurately, by GPA) is a kit consisting of locks and associated parts, appropriately coloured, coded and accompanied by instructions for a particular ship, or class of ships. Each kit, when complete and ready for despatch, measures about 1.1 x 0.5 x 0.5 m. The individual parts of the kit are made by manufacturers, but the applicant himself undertakes the final assembly, at times with the assistance of his wife. He does not employ any other person. Compendiously, the work involved in the “Lockout” business is the receipt of orders from customers, the ordering and receipt of component parts of the kits, the assembly of the kits, the despatch of the kits and the invoicing. The applicant accepted that, while he was occupied in these tasks, he could not be carrying out his consulting work. However, it seems that the time intervals involved in the “Lockout” business were such as to permit the applicant a degree of discretion with respect to when he would physically carry out the work involved in that business. He was asked about these matters, and he maintained the position that the operation of the business would not have compromised his ability to undertake consulting work about 25% more than he did (ie for about 100%, instead of about 80%, of the time) had he been in completely good health. There was nothing obviously implausible about the applicant’s position in this regard, and he was cross-examined only (as indicated in this paragraph) by drawing his attention to some of the practical aspects of the “Lockout” business which would require his attention. Valid though the respondents’ points were conceptually, I am unable to second-guess the applicant’s own estimation of what he would have been able to do absent his heart attack, and I am not persuaded that it has been undermined.
1003 The respondents next took issue with the applicant’s claim that his heart attack reduced his ability to take on consulting jobs which required extensive travel. They referred to invoices which showed that the applicant had travelled to quite distant places overseas, including Japan and Dubai, and that he undertook regular interstate travel, including travel to remote mine sites on occasion. They referred to the evidence of Mr Gibb that he unilaterally assessed the appropriateness of a particular job for the applicant, only then conferring with the applicant as to his readiness to undertake the job in question. The respondents submitted that the evidence demonstrates that the applicant is well able to undertake most of the assignments which Mr Gibb would be disposed to give him, and that, in the result, any diminution in the amount of work which he carries out − in comparison to a notional full time program − is unrelated to his heart attack. However, Mr Gibb made it clear that there was an element of discretion exercised by him (on account of the applicant’s heart condition) in the assignment of work to the applicant. Mr Gibb would not assign the applicant to locations which lacked the appropriate medical facilities, nor for jobs which would have required the applicant to undertake arduous travel.
1004 There does not seem to be any real doubt but that the applicant’s heart attack has limited the range of work assignments that he would otherwise be able to undertake. For that reason, the applicant has limited himself to about 8-10 days per month. He was not cross-examined about the appropriateness of this limitation. It was not directly suggested to him that, if the work were available, he could exceed this limitation. It was not put to him that the limitation was an unreasonably conservative response on his part to the fact that he had had a heart attack. In my view, although a reluctance to travel to inappropriate destinations may be part of the story, the effective limitation upon the applicant’s work is the 8-10 day one to which I have referred. I do not, therefore, think that much turns on the question whether the applicant might be able to undertake more distant assignments than he presently concedes.
1005 The respondents’ final point is that Mr Thompson had made no allowance for the global financial crisis. They went, however, no further than to make a submission in those terms. I am not prepared to qualify what would otherwise be my conclusion in relevant respects by reference to this consideration.
1006 For the above reasons, I accept so much of the respondents’ case as proposed that the growth figures for SWS provided by Mr Gibb should stand as a proxy for the extent to which the applicant’s consulting income would have grown between January 2004 and June 2008. I do not accept their attack on the applicant’s estimate that his heart attack compromised his ability to earn that income by about 20%. It follows that the figure which I would accept as representing the applicant’s economic loss over that period is $84,354.
1007 Mr Thompson also calculated a figure representing the future economic loss which he estimated would be suffered by the applicant. He did this using the present value (as of 28 February 2009) of the same weekly loss of income as he had estimated for the period 1 July 2008 − 28 February 2009. He assumed that the applicant would have worked until his 65th birthday and he reduced the figure by 15% for “contingencies”. His estimate was $84,788. Subject to the matters with which I have already dealt, the respondents accepted that estimate. I shall do likewise.
1008 It was submitted on behalf of the applicant that the assumption that he would, in the absence of his heart attack, have worked only to the age of 65 was a conservative one. This submission was based on evidence by the applicant in his witness statement that he never had a retirement date in mind, that he wanted to work as long as possible, and that retirement was “a very unappealing idea” to him. Counsel submitted that the applicant “might reasonably be allowed another two years’ loss of capacity to earn”. There was, however, no basis in the evidence for an allowance of that particular order (ie rather than of some other order). Indeed, ultimately counsel did not invite me to depart from Mr Thompson’s calculation as to future economic loss, contenting themselves with proposing that, if the court were to consider that there were factors raised by the respondents that might otherwise have an impact on the “economic loss outcome”, those factors might be “balanced against such considerations as the conservative assumption of only 20% loss … and the conservative assumption of age 65 retirement instead of 67 or older”. However, the only respect in which I have not accepted the applicant’s case on economic loss is one which is unrelated to the future. It would, therefore, be inappropriate to qualify the conclusion which I reached above in that respect by reference to a view (which, as it happens, I do not entertain) about the likelihood of the applicant working beyond the age of 65.
1009 The applicant’s heart attack will make it necessary for him to take medications into the future, and to undergo a range of regular medical tests and examinations. Prof Harper gave evidence of these requirements, and of the approximate anticipated cost of them. In final submissions made on his behalf, the applicant calculated that the present value of the costs associated with these requirements over the applicant’s expected life, discounted by 15% for contingencies, was $33,770. The only respect in which the respondents challenged this figure was to resist the inclusion of three drugs required to be taken by the applicant which, as Prof Harper agreed in his oral evidence, ought to have been taken by him in any event: aspirin, Coversyl and Lipitor. In final submissions, the applicant did not deal with this submission of the respondents. I accept the submission. The figure of $33,770 should be re-calculated to exclude the cost of the three drugs referred to.
1010 The applicant seeks the inclusion in damages of the sum of $4,175.35 said to be payable by him pursuant to a Notice of Charge, dated 20 April 2009, under s 8 of the Health and Other Services (Compensation) Act 1995 (Cth). The respondents accepted that the applicant had produced the notice to them, but added: “[h]owever it is not in evidence”. There appears to be substance in that unworthy submission. As things stand, I could not allow the applicant this modest amount, but I do not exclude the possibility that the applicant might seek leave to tender the notice when the proceeding is listed to settle the orders to be made.
1011 The applicant sought damages for “the costs associated with any future cardiovascular event causally related to this myocardial infarction in accordance with the risk of future occurrence of such an event”, relying in this regard upon Malec v JC Hutton Pty Ltd (1990) 169 CLR 638. However, the evidence would not sustain the conclusion that the heart attack which the applicant had in December 2003 has made it more likely that he will have another one: see para 990 above. For that reason, I reject this lead of damages.
1012 The applicant sought damages in respect of a contingent need for attendant care should he suffer “heart failure” in the future, even in the absence of a heart attack. This claim was based on the following paragraphs in Prof Harper’s report:
Mr Peterson currently has about a 5% to 10% chance of a further heart attack over the next five years. A subsequent heart attack would be likely to cause further damage to his heart muscle and because of the damage his heart muscle has already sustained as a result of his heart attack in 2003, there is arisk that a further heart attack would cause heart failure sooner rather than later.
Heart failure is a condition where the heart does not supply sufficient oxygenated blood to meet the needs of the body. This means that daily activities become extremely difficult to perform, as the smallest exertion can
place an unacceptable burden on the heart.
If Mr Peterson went into heart failure his future medical needs would be likely to involve more intensive drug therapy, more frequent visits to both his cardiologist and his general practitioner. He may require rehabilitation. He may require attendant care. The degree of attendant care would depend on the severity of the heart failure. The 5 year survival prognosis for patients with heart failure is
50%.
With the passage of time, Mr Peterson may develop heart failure even in the absence of further heart attacks. Such a scenario would be unlikely to occur within the next 15 years. If Mr Peterson developed heart failure without further heart attack, his needs would be as described in paragraph 61 above.
From this the applicant submitted that, although his risk of a further heart attack in the next five years was only 5% − 10%, if that did occur it would likely have consequences that required the engagement of attendant care. So much may be accepted, but it seems very problematic, and has not been satisfactorily established, that the events of December 2003 have on balance made it more likely that the applicant would at some time in the future require attendant care than would otherwise have been the case.
1013 Neither was the applicant’s quantification of this head of damage satisfactory. There was no evidence about the cost of attendant care. The applicant proposed that I should “assess a sum of $100,000”, and then discount that sum by 50% to take account of various uncertainties and contingencies to which he referred in his submissions. Those submissions were, however, almost entirely intuitive. There was neither evidence nor submission to explain why $100,000, rather than some other sum, should be assessed. Neither did the applicant explain why some evidence on this subject had not been called. For my own part, it does not seem at all obvious that some short evidence about the nature and cost of the kind of attendant care typically required by people who had suffered heart failure might not have been called. The respondents resisted the applicant’s claim under this head upon the ground (amongst others) that there was no “detailed basis” for the figure claimed. For reasons set out above, I am bound to uphold their position. I reject this head of damages.
1014 I shall require the applicant to bring in minutes of orders appropriate to give effect to my determination his claim for compensation under ss 74B and 74D of the Trade Practices Act, conformably with my reasons set out above.
|
I certify that the preceding One thousand and fourteen (1014) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Jessup. |
Associate:
Dated: 5 March 2010
appendix A
IN THE FEDERAL COURT OF AUSTRALIA
VICTORIA DISTRICT REGISTRY
No: (P)VID451/2006
GRAEME ROBERT PETERSON
Plaintiff
MERCK SHARPE & DOHME (AUST) PTY LTD
First Defendant
MERCK & CO INC
Second Defendant
ORDER
|
JUDGE:
|
Justice Jessup |
|
DATE OF ORDER:
|
30 March 2009 |
|
WHERE MADE:
|
Melbourne |
THE COURT ORDERS THAT:
1. Pursuant to section 33ZF of the Federal Court of Australia Act 1976, that at the hearing to commence on 30 March 2009, in advance of all other issues in the proceeding, the following issues are to be determined:
a. the issues of fact and law in the claim brought by Mr Peterson in his personal capacity; and
b. those issues of fact and law set out in the Schedule attached to this Order that the Court finds to be common to the claims of the group members.
Date that entry is stamped: 15 June 2009
Deputy District Registrar
|
IN THE Federal Court OF AUSTRALIA Victoria District Registry |
|
No. VID 451 of 2009
|
GRAEME ROBERT PETERSON
Applicant
-and-
Merck Sharp & Dohme (Australia) Pty Ltd
ACN 000 173 508
First Respondent
MERCK & CO., INC.
Second Respondent
SCHEDULE
Definitions
In this Schedule:
Health care professionals includes medical practitioners, pharmacists and other health care professionals.
Vioxx cardiovascular conditions means each of myocardial infarction; thrombotic stroke; unstable angina; transient ischaemic attack; and peripheral vascular disease.
Vioxx includes a reference to Vioxx tablets and rofecoxib.
Commonality
1. Are any and which of the questions set out below common to the claims of the group members?
Materially increased risk
2. Is there a physiological mechanism by which the consumption of Vioxx is capable of causing any and which of the Vioxx cardiovascular conditions?
3. If the answer to question 2 is in the affirmative, then for each such condition, what is the physiological mechanism?
4. Did the consumption of Vioxx increase the risk of suffering any and which of the Vioxx cardiovascular conditions?
5. If the answer to question 4 is in the affirmative, was the increase in risk material?
Negligence
6. If the answer to question 5 is in the affirmative:
(a) when, if ever, did the First Respondent (“Merck Australia”) first know that the consumption of Vioxx materially increased the risk of suffering the condition?
(b) when, if ever, ought Merck Australia have first known that that consumption of Vioxx materially increased the risk of suffering the condition?
(c) did Merck Australia owe the group members a duty to take reasonable care to avoid acts and omissions that may expose them to a material increase in the risk of suffering the condition as a consequence of consuming Vioxx tablets?
7. Did Merck Australia:
(a) not owe group members the duty of care alleged; or
(b) satisfy the applicable standard of care
by virtue of compliance with the relevant legislative requirements referred to in paragraph 61 (a)-(t) of the Defence to Further Amended Statement of Claim and requirements imposed upon it in respect of Vioxx tablets by the Therapeutic Goods Administration (“TGA”) pursuant to the powers granted by the Therapeutic Goods Act 1989 (Cth)?
8. Did Merck Australia:
(a) not owe group members the duty of care alleged; or
(b) satisfy the applicable standard of care
by reason of the learned intermediary defence pleaded in paragraph 63 of the Defence to Further Amended Statement of Claim?
9. If the answer to question 5 is in the affirmative:
(a) did Merck Australia fail to make adequate inquiries of the Second Respondent (“Merck Inc”) regarding the adverse side effects and health risks that may be associated with the consumption of Vioxx including a material increase in the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(b) Did Merck Australia fail to undertake or cause to be undertaken any or any adequate research, investigations, clinical trials or observational studies in order to ascertain the adverse side effects and health risks that may be associated with the consumption of Vioxx including a material increase in the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(c) Did Merck Australia fail to have adequate regard to the results of any research, investigations, clinical trials or observational studies undertaken or caused to be undertaken by Merck Australia, Merck Inc or others suggesting that the consumption of Vioxx materially increased the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(d) Did Merck Australia fail to provide adequate information, advice or warning to the general public or to health care professionals to the effect that the consumption of Vioxx materially increased the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(e) Did Merck Australia develop and implement a marketing strategy or campaign by which it formulated and disseminated to health care professionals in Australia any and which of the following representations about Vioxx:
(i) Vioxx has an excellent gastrointestinal safety profile compared with other non-steroidal anti-inflammatory drugs (“NSAIDs”);
(ii) Vioxx has an excellent overall safety profile compared with other NSAIDs;
(iii) Vioxx does not increase the incidence of adverse cardiovascular events;
(iv) higher rates of myocardial infarction and other cardiovascular events among persons taking Vioxx relative to persons taking other traditional NSAIDs (in particular naproxen) did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events but were attributable to the fact that the consumption of naproxen produced a lower incidence of cardiovascular events because naproxen inhibits platelet aggregation in a manner similar to aspirin while Vioxx does not;
(v) the higher rates of myocardial infarction among persons taking Vioxx relative to persons taking naproxen observed in the Vioxx Gastrointestinal Outcomes Research Trial (“Vigor”) did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events;
(vi) any information to the effect that Vioxx was not as safe as other NSAIDs because the consumption of Vioxx materially increased the risk of suffering the condition was incorrect, misleading, exaggerated and/or unreliable.
If so, was that conduct less than reasonable in all of the circumstances?
10. Was it less than reasonable in all of the circumstances, at any time before 30 September 2004, for Merck Australia to distribute Vioxx in Australia? If so, when?
11. Is the fact that the Product Information for Vioxx was approved by the TGApursuant to the Therapeutic Goods Act , if proved, a complete answer to an allegation that Merck Australia was in breach of any common law duty by unreasonably failing to adequately disclose information in the Product Information?
Section 52 of the Trade Practices Act 1974 (Cth)
12. If the answer to question 5 is in the affirmative, did Merck Australia:
(a) label Vioxx without providing adequate information, advice or warning to the general public or to health care professionals to the effect that the consumption of Vioxx materially increased the risk of suffering the condition?
(b) market and distribute and/or supply Vioxx for sale in Australia without providing adequate information, advice or warning to the general public or to health care professionals to the effect that the consumption of Vioxx materially increased the risk of suffering the condition?
(c) develop and implement a marketing strategy or campaign by which it formulated and disseminated to health care professionals in Australia, any and which of the following representations about Vioxx:
(i) Vioxx has an excellent gastrointestinal safety profile compared with other NSAIDs;
(ii) Vioxx has an excellent overall safety profile compared with other NSAIDs;
(iii) Vioxx does not increase the incidence of adverse cardiovascular events;
(iv) higher rates of myocardial infarction and other cardiovascular events among persons taking Vioxx relative to persons taking other traditional NSAIDs (in particular naproxen) did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events but were attributable to the fact that the consumption of naproxen produced a lower incidence of cardiovascular events because naproxen inhibits platelet aggregation in a manner similar to aspirin while Vioxx does not;
(v) the higher rates of myocardial infarction among persons taking Vioxx relative to persons taking naproxen observed in Vigor did not indicate that Vioxx increased the risk of suffering adverse cardiovascular events;
(vi) information to the effect that Vioxx was not as safe as other NSAIDs because the consumption of Vioxx materially increased the risk of suffering the condition was incorrect, misleading, exaggerated and/or unreliable?
13. If Merck engaged in any of the conduct referred to in question 12 was any of that conduct in trade or commerce within the meaning of the Trade Practices Act ? If so, which conduct?
14. Was any of the conduct identified in question 12, in all of the circumstances and context in which it occurred, misleading or deceptive or likely to mislead or deceive in contravention of section 52 of the Trade Practices Act ?
15. Upon the proper construction of the Therapeutic Goods Act , does the fact that the Product Information for Vioxx was approved by the TGA pursuant to the Therapeutic Goods Act , if proved, exclude the operation of the Trade Practices Act in so far as an allegation is made that a failure to adequately disclose information in the Product Information contravenes:
(a) section 52 of the Trade Practices Act?
(b) section 75AD of the Trade Practices Act?
16. Is the fact that Vioxx was registered on the Australian Register of Therapeutic Goods pursuant to the Therapeutic Goods Act and available only by prescription of permitted prescribers, and that the Product Information for Vioxx was approved by the TGApursuant to the Therapeutic Goods Act, if proved, a complete answer to an allegation that the respondent contravened:
(a) section 52 of the Trade Practices Act;
(b) section 75AD of the Trade Practices Act;
by failing to adequately disclose information in the Product Information?
Sections 75AD, 74B and 74D of the Trade Practices Act 1974 (Cth)
17. Were Vioxx tablets manufactured by Merck Australia “goods” that were "ordinarily acquired" as defined in section 74A(2) of the Trade Practices Act?
18. Is the fact that Vioxx was registered on the Australian Register of Therapeutic Goods pursuant to the Therapeutic Goods Act andavailable in its approved form and packaging only by prescription of permitted prescribers, if proved, sufficient to establish as a matter of law that Vioxx was reasonably fit for purpose within the meaning of s74B of the Trade Practices Act ?
19. Is the fact that Vioxx was registered on the Australian Register of Therapeutic Goods pursuant to the Therapeutic Goods Act and available in its approved form and packaging only by prescription of permitted prescribers, if proved, sufficient to establish as a matter of law that Vioxx was of merchantable quality within the meaning of s74D of the Trade Practices Act ?
20. If the answer to question 5 is in the affirmative, were Vioxx tablets defective within the meaning of section 75AD of the Trade Practices Act in that the safety of Vioxx tablets was not such as persons are generally entitled to expect by reason of the fact that the consumption of Vioxx materially increased the risk of suffering the condition?
21. If the answer to question 5 is in the affirmative, were Vioxx tablets defective within the meaning of section 75AD of the Trade Practices Act in that the safety of Vioxx was not such as persons are generally entitled to expect by reason of the fact that the packaging and labelling of Vioxx and the Vioxx product information did not contain adequate information, advice or warning to the effect that the consumption of Vioxx materially increased the risk of suffering the condition?
22. If the answer to question 5 is in the affirmative, were Vioxx tablets not reasonably fit for the purpose of acquisition within the meaning of section 74B of the Trade Practices Act by reason of the fact that the consumption of Vioxx materially increased the risk of suffering the condition?
23. If the answer to question 5 is in the affirmative, were Vioxx tablets not of merchantable quality within the meaning of section 74D of the Trade Practices Act by reason of the fact that the consumption of Vioxx materially increased the risk of suffering the condition?
24. If the answer to question 5 is in the affirmative, at all times before 30 September 2004 was the state of scientific or technical knowledge such as to enable Merck Australia to discover that the consumption of Vioxx materially increased the risk of suffering the condition?
25. Were the defects pleaded in paragraphs 33, 34 and 37 of the Further Amended Statement of Claim (if found to exist) caused by Merck Australia’s compliance with a mandatory standard or standards?
Claim against Merck Inc
26. If the answer to question 5 is in the affirmative:
(a) when did Merck Inc first know that the consumption of Vioxx materially increased the risk of suffering the condition?
(b) when ought Merck Inc to have first known that the consumption of Vioxx materially increased the risk of suffering the condition?
(c) did Merck Inc owe the group members a duty to take reasonable care to avoid acts and omissions that may expose them to a material increase in the risk of suffering the condition as a consequence of consuming Vioxx?
27. Did Merck Inc:
(a) not owe group members the duty of care alleged; or
(b) satisfy the applicable standard of care
by virtue of Merck Australia’s compliance with the relevant legislative requirements referred to in paragraphs 23(a)-(1) of the Defence to Amended Statement of Claim and the requirements imposed upon Merck Australia by the TGA pursuant to the powers granted by the Therapeutic Goods Act?
28. Did Merck Inc:
(a) not owe group members the duty of care alleged; or
(b) satisfy the applicable standard of care
by reason of the learned intermediary defence pleaded in paragraph 24 of the Defence to the Further Amended Statement of Claim?
29. If the answer to question 5 is in the affirmative:
(a) did Merck Inc fail to undertake or cause to be undertaken any or any adequate research, investigations, clinical trials or observational studies in order to ascertain the adverse side effects and health risks that may be associated with the consumption of Vioxx including a material increase in the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(b) did Merck Inc fail to have adequate regard to the results of any research, investigations, clinical trials or observational studies undertaken or caused to be undertaken by Merck Inc or others suggesting that the consumption of Vioxx materially increased the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
(c) Did Merck Inc withhold from and fail to publish in medical publications or otherwise reveal data and information of which it was aware concerning adverse cardiovascular risks associated with the consumption of Vioxx? If so, was that failure less than reasonable in all of the circumstances?
(d) Did Merck Inc fail to provide to Merck Australia, and health care professionals in Australia or the Australian public any or any adequate information, advice or warning to the effect that the consumption of Vioxx materially increased the risk of suffering the condition? If so, was that failure less than reasonable in all of the circumstances?
30. Was it less than reasonable in all of the circumstances at any time before 30 September 2004 for Merck Inc to supply Vioxx to Merck Australia in Australia? If so, when?
APPENDIX B
ADAPT Research Group. Cardiovascular and Cerebrovascular Events in the Randomized, Controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials 2006;1(7):e33.
Antman EM, et al. Cyclooxygenase Inhibition and Cardiovascular Risk. Circulation 2005; 112: 759-70.
APTC (Antiplatelet Trialists’ Collaboration). Collaborative overview of randomised trials of antiplatelet therapy-I: prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. BMJ 1994;308:81-106.
Arber N, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med 2006;355(9):885-95.
ATT (Antithrombotic Trialists’ Collaboration). Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002;324:71-86.
Aw T, et al. Meta-analysis of Cyclooxygenase-2 Inhibitors and Their Effects on Blood Pressure. Arch Intern Med 2005;165:490-6.
Baigent C, Patrono C. Selective Cyclooxygenase 2 Inhibitors, Aspirin, and Cardiovascular Disease. Arthritis Rheum 2003;48(1): 12-20.
Baron JA, et al. A Randomized Trial of Aspirin to Prevent Colorectal Adenomas. N Engl J Med 2003;348:891-9.
Baron JA, et al. Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial. Lancet 2008;372:1756-64.
Bombardier C, et al. Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis. N Engl J Med 2000;343:1520-8.
Bresalier RS, et al. Cardiovascular Events Associated with Rofecoxib in a Colorectal Adenoma Chemoprevention Trial. N Engl J Med 2005;352:1092-1102.
Bresalier RS, et al. Correction. N Engl J Med 2006;355(2):221.
Brochier ML. Evaluation of flurbiprofen for prevention of reinfarction and reocclusion after successful thrombolysis of angioplasty in acute myocardial infarction. Eur Heart J 1993;14:951-7.
Capone ML, et al. Clinical Pharmacology of Platelet, Monocyte, and Vascular Cyclooxygenase Inhibition by Naproxen and Low-Dose Aspirin in Healthy Subjects. Circulation 2004;109:1468-71.
Catella-Lawson F, et al. Effects of Specific Inhibition of Cyclooxygenase-2 on Sodium Balance, Hemodynamics and Vasoactive Eicosanoids. J Pharmacol Exp Ther 1999;289(2):735-41.
Chen L, Ashcroft DM. Risk of myocardial infarction associated with selective COX-2 inhibitors: Meta-analysis of randomised controlled trials. Pharmacoepidemiology Drug Safety 2007;16(7):762-72.
Clelend LG, et al. COX-2 inhibition and thrombotic tendency: a need for surveillance. Med J Aust 2001;175:214-7.
Curfman GD, et al. Expression of Concern: Bombardier et al., “Comparison of Upper Gastrointestinal Toxicity of Rofecoxib and Naproxen in Patients with Rheumatoid Arthritis,” N Engl J Med 2000;343:1520-8. N Engl J Med 2005;353(26):2813-4.
Daniels B, Seidenberg B. Cardiovascular Safety Profile of Rofecoxib in Controlled Clinical Trials. Arthritis Rheum 1999;Suppl(435):S143.
Dekker RJ, et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 2002;100(5):1689-98.
Egan KM, et al. Cyclooxygenases, Thromboxane, and Atherosclerosis: Plaque Destabilization by Cyclooxygenase-2 Inhibition Combined With Thromboxane Receptor Antagonism. Circulation 2005;111:334-42.
Farkouh ME, et al. Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomized controlled trial. Lancet 2004;364:675-84.
FitzGerald GA, Patrono C. The Coxibs, Selective Inhibitors of Cyclooxygenase-2. N Engl J Med 2001;345(6):433-42.
Fornaro G, et al. Indobufen in the Prevention of Thromboembolic Complications in Patients With Heart Disease A Randomized, Placebo-Controlled, Double-Blind Study. Circulation 1993;87(1):162-4.
Garcia Rodriguez LA, et al. Nonsteroidal Antiinflammatory Drugs and the Risk of Myocardial Infarction in the General Population. Circulation 2004;109:3000-6.
Graham DJ, et al. Risk of acute myocardial infarction and sudden cardiac death in patients treated with cyclo-oxygenase 2 selective and non-selective non-steroidal anti-inflammatory drugs: nested case-control study. Lancet 2005;365(9458):475-81.
Grosser T, et al. Biological basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest 2006;116(1):4-15.
Hill AB. The Environment and Disease: Association or Cause? Proc R Soc Med 1965;295-300.
Hippisley-Cox J, Coupland C. Risk of myocardial infarction in patients taking cyclo-oxygenase-2 inhibitors or conventional non-steroidal anti-inflammatory drugs: population based nested case-control analysis. BMJ 2005;330:1366-72
Hla T, Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci USA 1992;89:7384-8.
Jaffe EA, et al. Recovery of Endothelial Cell Prostacyclin Production after Inhibition by Low Doses of Aspirin. J Clin Invest 1979;63:532-5.
Jick SS. The Risk of Myocardial Infarction, and Newly Diagnosed Hypertension in Users of Meloxicam, Diclofenac, Naproxen, and Piroxicam. Pharmacotherapy 2000;20(7):741-4.
Johnsen SP, et al. Risk of Hospitilization for Myocardial Infarction Among Users of Rofecoxib, Celecoxib, and Other NSAIDs. Arch Intern Med 2005;165:978-84.
Jones DA, et al. Molecular Cloning of Human Prostaglandin Endoperoxide Synthase Type II and Demonstration of Expression in Response to Cytokines. J Biol Chem 1993;268(12):9049-54.
Jüni P, et al. Risk of cardiovascular events and rofecoxib: cumulative meta-analysis. Lancet 2004;364(9450):2021-9.
Kearney PM, et al. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. BMJ 2006;332:1302-8.
Kimmel SE, et al. The Effects of Nonselective Non-Aspirin Non-Steroidal Anti-Inflammatory Medications on the Risk of Nonfatal Myocardial Infarction and Their Interaction With Aspirin. J Am Coll Cardiol 2004:43:985-90.
Konstam MA, et al. Cardiovascular Thrombotic Events in Controlled, Clinical Trials of Rofecoxib. Circulation 2001;104:2280-8.
Kuklinska AM, et al. Low dose rofecoxib, Inflammation and prostacyclin synthesis in acute coronary syndromes. Rocz Akad Med Bialymst 2005;50:339-42.
Lagakos SW. Time-to-event Analyses for Long-Term Treatments—The APPROVe Trial. N Engl J Med 2006;355(2):113-7.
Lekakis JP, et al. Divergent effects of rofecoxib on endothelial function and inflammation in acute coronary syndromes. Int J Cardiol 2006;112(3):359-66.
Lévesque LE, et al. The Risk of Myocardial Infarction with Cyclooxygenase-2 Inhibitors: A Population Study of Elderly Adults. Ann Intern Med 2005;142:481-9.
Lichtenstein DR, Wolfe MM. COX-2 Selective NSAIDs. New and Improved? JAMA 2000;284:1297-9.
Lisse JR, et al. Gastrointestinal Tolerability and Effectiveness of Rofecoxib versus Naproxen in the Treatment of Osteoarthritis. Ann Intern Med 2003;139:539-46.
Mamdani M, et al. Effect of Selective Cyclooxygenase 2 Inhibitors and Naproxen on Short-term Risk of Acute Myocardial Infarction in the Elderly. Arch Intern Med 2003;163:481-6.
McAdam BF, et al. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: The human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci USA 1999;96:272-7.
McAdam BF, et al. Contribution of Cyclooxygenase-2 to Elevated Biosynthesis of Thromboxane A2 and Prostacyclin in Cigarette Smokers. Circulation 2005;112(7):1024-9.
McGettigan P, Henry D. Cardiovascular Risk and Inhibition of Cyclooxygenase: A Systematic Review of the Observational Studies of Selective and Nonselective Inhibitors of Cyclooxygenase 2. JAMA 2006;296(13):1633-44.
Mukherjee D, et al. Risk of Cardiovascular Events Associated With Selective COX-2 Inhibitors. JAMA 2001;286(8):254-9.
Nelson L and Cox M, Lehninger’s Principles of Biochemistry, 5th ed, 2008.
Nussmeier NA, et al. Complications of the COX-2 Inhibitors Parecoxib and
Valdecoxib after Cardiac Surgery. N Engl J Med 2005;352(11):1081-91.
Patrono C, et al. Platelet-Active Drugs: The Relationships Among Dose, Effectiveness, and Side Effects. Chest 2004;126:234S-264S.
Rahme E, et al. Association Between Naproxen Use and Protection Against Acute Myocardial Infarction. Arch Intern Med 2002;162:1111-5.
Rahme E, Nedjar H. Risks and benefits of COX-2 inhibitors vs non-selective NSAIDS: does their cardiovascular risk exceed their gastrointestinal benefit? A retrospective cohort study. Rheumatology 2007;46:435-8.
Ray WA, et al. COX-2 selective non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease. Lancet 2002;360(9339):1071-3.
Ray WA, et al. Non-steroidal anti-inflammatory drugs and risk of serious coronary heart disease: an observational cohort study. Lancet 2002;359(9301):118-23.
Reicin AS, et al. Comparison of Cardiovascular Thrombotic Events in Patients With Osteoarthritis Treated With Rofecoxib Versus Nonselective Nonsteroidal Anti-inflammatory Drugs (Ibuprofen, Diclofenac, and Nabumetone). Am J Cardiol 2002;89:204-9.
Ridker PM, et al. Inflammation, Aspirin, and the Risk of Cardiovascular Disease in Apparently Healthy Men. N Engl J Med 1997;336(14):973-9.
Ridker PM, et al. C-Reactive Protein Adds to the Predictive Value of Total and HDL Cholesterol in Determining Risk of First Myocardial Infarction. Circulation 1998;97:2007-11.
Ridker PM, et al. C-Reactive Protein and Other Markers of Inflammation in the Prediction of Cardiovascular Disease in Women. N Engl J Med 2000;342(12):836-43.
Rodriguez L, et al. Differential effects of aspirin and non-aspirin nonsteroidal antiinflammatory drugs in the primary prevention of myocardial infarction in postmenopausal women. Epidemiology 2000;11:382-7.
Sanmuganathan PS, et al. Aspirin for primary prevention of coronary heart disease: safety and absolute benefit related to coronary risk derived from meta-analysis of randomised trials. Heart 2001;85:265-71.
Schlienger RG, et al. Use of nonsteroidal anti-inflammatory drugs and the risk of first-time acute myocardial infarction. Br Clin Pharmacol 2002;54:327-32.
Scott PA, et al. Non-steroidal anti-inflammatory drugs and myocardial infarctions: comparative systematic review of evidence from observational studies and randomised controlled trials. Ann Reum Dis 2007;66:1296-304.
Solomon DH, et al. Nonsteroidal Anti-inflammatory Drug Use and Acute Myocardial Infarction. Arch Intern Med 2002;162:1099-104
Solomon SD, et al. Cardiovascular Risk Associated with Celecoxib in a Clinical Trial for Colorectal Adenoma Prevention. N Engl J Med 2005;352(11):1071-80.
Solomon SD, et al. Effect of Celecoxib on Cardiovascular Events and Blood Pressure in Two Trials for the Prevention of Colorectal Adenomas. Circulation 2006;114:1028-1035.
Thal LJ, et al. A Randomized, Double-Blind, Study of Rofecoxib in Patients with Mild Cognitive Impairment. Neuropsychopharmacology 2005;30:1204-15.
Tuleja E, et al. Effects of Cyclooxygenases Inhibitors on Vasoactive Prostanoids and Thrombin Generation at the Site of Microvascular Injury in Healthy Men. Arterioscler Thromb Vasc Biol 2003;23(6):1111-5.
Topper JN, et al. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: Cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci USA 1996;93:10417-22.
Tuleja E, et al. Effects of Cyclooxygenases Inhibitors on Vasoactive Prostanoids and Thrombin Generation at the Site of Microvascular Injury in Healthy Men. Arterioscler Thromb Vasc Biol 2003;23:1111-5.
Van Hecken A, et al. Comparative Inhibitory Activity of Rofecoxib, Meloxicam, Diclofenac, Ibuprofen, and Naproxen on COX-2 versus COX-1 in Healthy Volunteers. J Clin Pharmacol 2000;40:1109-20.
Walter MF, et al. Sulfone COX-2 Inhibitors increase susceptibly of human LDL and plasma to oxidative modification: comparison sulfonamide COX-2 inhibitors and NSAIDs. Atherosclerosis 2004;177(2):235-43.
Watson DJ, et al. Lower Risk of Thromboembolic Cardiovascular Events With Naproxen Among Patients With Rheumatoid Arthritis. Arch Intern Med 2002;162:1105-10.
Weir MR, et al. Selective COX-2 inhibition and cardiovascular effects: A review of the rofecoxib development program. Am Heart J 2003;146:591-604.
White WB, et al. Cardiovascular Thrombotic Events in Arthritis Trials of the Cyclooxygenase-2 Inhibitor Celecoxib. Am J Cardiol 2003;92:411-8.
Wong E, et al. Effects of COX-2 inhibitors on aortic prostacyclin production in cholesterol-fed rabbits. Atherosclerosis 2001;157(2):393-402.

