
FEDERAL COURT OF AUSTRALIA
Arrow Pharmaceuticals Limited v Merck & Co., Inc. [2004] FCA 1282
PATENTS – application for revocation – attempt to ‘evergreen’ pharmaceutical patent – validity – whether new dosing regime for administering known substance for known purpose by known method without new pharmaceutical properties constitutes a manner of manufacture or an invention
PATENTS – application for revocation – novelty – whether claimed invention anticipated by information in commercial magazine – magazine distributed internationally to existing and potential customers of commercial corporation – recipients of magazine free in law and equity to make use of information contained therein – no evidence magazine available on request or for inspection in any public library – evidence of publication of contents of magazine via the internet – whether information ‘publicly available’ so as to constitute ‘prior art information’ for the purposes of the Patents Act 1990 (Cth)
PATENTS – application for revocation – novelty – dosage regime for administering alendronate – whether method anticipated by prior art information – application of reverse infringement test – prior art information suggested testing of claimed invention – whether suggestion that disclosed invention be tested prevented disclosure from constituting anticipation – whether suggestion of specific doses anticipated claimed invention
PATENTS – application for revocation – false suggestion – patent specification contained statements alleged to misrepresent conclusions drawn from published article – no finding that any such misrepresentation intended to mislead – whether patent obtained by fraud, false suggestion or misrepresentation
WORDS AND PHRASES – ‘made publicly available’
Patents Act 1990 (Cth), ss 7, 18, 40, 80–87, 138
Adhesive Dry Mounting Co Ltd v Trapp & Co (1910) 27 RPC 341 referred to
Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd (1998) 194 CLR 171 considered
Anaesthetic Supplies Pty Ltd v Rescare Ltd (1994) 50 FCR 1 followed
Azuko Pty Ltd v Old Digger Pty Ltd (2001) 52 IPR 75 cited
Bristol-Myers Squibb Co v Baker Norton Pharmaceuticals Inc [2001] RPC 1 referred to
Bristol-Myers Squibb Co v FH Faulding & Co Ltd (2000) 97 FCR 524 followed in part; distinguished in part
Commissioner of Patents v Microcell Ltd (1959) 102 CLR 232 applied
Fomento Industrial SA v Mentmore Manufacturing Co Ltd [1956] RPC 87 cited
ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc (2000) 106 FCR 214 cited
Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 cited
Merck & Co., Inc’s Patents [2003] EWCA Civ 1545; [2004] FSR 16 referred to
Merck & Co., Inc’s Patents [2003] EWHC 5 (Pat); [2003] FSR 29 referred to
Meyers Taylor Pty Ltd v Vicarr Industries Ltd (1977) 137 CLR 228 applied
National Research Development Corporation v Commissioner of Patents (1959) 102 CLR 252 considered
NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 applied
Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 applied
PLG Research Ltd v Ardon International Ltd [1993] FSR 197 cited
Re Bristol-Myers Co’s Application [1969] RPC 146 cited
Sunbeam Corporation v Morphy-Richards (Australia) Pty Ltd (1961) 180 CLR 98 followed
Telstra Corporation Limited v Australasian Performing Right Association Limited (1997) 191 CLR 140 cited
Wellcome Foundation Ltd v Commissioner of Patents (1980) 145 CLR 520 referred to
D Falconer, W Aldous & D Young, Terrell on the Law of Patents, 12th edn, Sweet & Maxwell, London, 1971
ARROW PHARMACEUTICALS LTD (ACN 088 417 403) v MERCK & CO., INC.
N 1211 OF 2002
GYLES J
6 OCTOBER 2004
SYDNEY
| IN THE FEDERAL COURT OF AUSTRALIA |
|
| NEW SOUTH WALES DISTRICT REGISTRY | N 1211 OF 2002 |
| BETWEEN: | ARROW PHARMACEUTICALS LIMITED (ACN 088 417 403) APPLICANT
|
| AND: | MERCK & CO., INC. RESPONDENT
|
| GYLES J | |
| DATE OF ORDER: | |
| WHERE MADE: | SYDNEY |
THE COURT ORDERS THAT:
The proceeding stand over for the making of orders.
Note: Settlement and entry of orders is dealt with in Order 36 of the Federal Court Rules.
| IN THE FEDERAL COURT OF AUSTRALIA |
|
| NEW SOUTH WALES DISTRICT REGISTRY | N 1211 OF 2002 |
| BETWEEN: | ARROW PHARMACEUTICALS LIMITED (ACN 088 417 403) APPLICANT
|
| AND: | MERCK & CO., INC. RESPONDENT
|
| JUDGE: | GYLES J |
| DATE: | |
| PLACE: | SYDNEY |
REASONS FOR JUDGMENT
1 This is an application by Arrow Pharmaceuticals Limited (Arrow) for revocation of a number of claims of a patent granted pursuant to the Patents Act 1990 (Cth) (the Act). The case involves what would now colloquially be called an attempt to ‘evergreen’ a pharmaceutical patent. I find that the application for revocation succeeds. The patent in suit (No 741818) is titled ‘Method for inhibiting bone resorption’. The patentee is the respondent Merck & Co., Inc. (Merck), an American corporation with a substantial business of supplying pharmaceutical products. The abstract of the invention is as follows:
‘Disclosed are methods for inhibiting bone resorption in mammals while minimizing the occurrence of or potential for adverse gastrointestinal effects. Also disclosed are pharmaceutical compositions and kits for carrying out the therapeutic methods disclosed herein.’
2 The field of the invention is described as follows:
‘The present invention relates to oral methods for inhibiting bone resorption in a mammal while minimizing the occurrence of or potential for adverse gastrointestinal effects. These methods comprise orally administering to a mammal in need thereof of a pharmaceutically effective amount of a bisphosphonate as a unit dosage according to a continuous schedule having a dosing interval selected from the group consisting of once-weekly dosing, twice-weekly dosing, biweekly dosing, and twice-monthly dosing. The present invention also relates to pharmaceutical compositions and kits useful for carrying out these methods.’
3 The background of the invention may be gleaned from the account of it in the specification which I will set out in more detail than normal because of the issues in the case. Relevant portions (omitting references) are as follows:
‘A variety of disorders in humans and other mammals involve or are associated with abnormal bone resorption. Such disorders include, but are not limited to, osteoporosis, Paget’s disease, periprosthetic bone loss or osteolysis, and hypercalcemia of malignancy. The most common of these disorders is osteoporosis, which in its most frequent manifestation occurs in postmenopausal women. Osteoporosis is a systemic skeletal disease characterized by a low bone mass and microarchitectural deterioration of bone tissue, with a consequent increase in bone fragility and susceptibility to fracture. Because osteoporosis, as well as other disorders associated with bone loss, are chronic conditions, it is believed that appropriate therapy will generally require chronic treatment.
Multinucleated cells called osteoclasts are responsible for causing bone loss through a process known as bone resorption. It is well known that bisphosphonates are selective inhibitors of osteoclastic bone resorption, making these compounds important therapeutic agents in the treatment or prevention of a variety of generalized or localized bone disorders caused by or associated with abnormal bone resorption. …
At present, a great amount of preclinical and clinical data exists for the potent bisphosphonate compound alendronate. Evidence suggests that other bisphosphonates such as risedronate, tiludronate, ibandronate and zolendronate, have many properties in common with alendronate, including high potency as inhibitors of osteoclastic bone resorption. An older bisphosphonate compound, etidronate, also inhibits bone resorption. However, unlike the more potent bisphosphonates, etidronate impairs mineralization at doses used clinically, and may give rise to osteomalacia, a condition resulting in an undesirable decrease in bone mineralization. …
Despite their therapeutic benefits, bisphosphonates are poorly absorbed from the gastrointestinal tract. … Intravenous administration has been used to overcome this bioavailability problem. However, intravenous administration is costly and inconvenient, especially when the patient must be given an intravenous infusion lasting several hours on repeated occasions.
If oral administration of the bisphosphonate is desired, relatively high doses must be administered to compensate for the low bioavailability from the gastrointestinal tract. To offset this low bioavailability, it is generally recommended that the patient take the bisphosphonate on an empty stomach and fast for at least 30 minutes afterwards. However, many patients find the need for such fasting on a daily basis to be inconvenient. Moreover, oral administration has been associated with adverse gastrointestinal effects, especially those relating to the esophagus. … These effects appear to be related to the irritant potential of the bisphosphonate in the esophagus, a problem which is exacerbated by the presence of refluxed gastric acid. For example, the bisphosphonate, pamidronate has been associated with esophageal ulcers. … Although not as common, the use of alendronate has been associated with esophagitis and/or esophageal ulcers. … The degree of adverse gastrointestinal effects of bisphosphonates has been shown to increase with increasing dose. … Also, these adverse esophageal effects appear to be more prevalent in patients who do not take the bisphosphonate with an adequate amount of liquid or who lie down shortly after dosing, thereby increasing the chance for esophageal reflux.
Current oral bisphosphonate therapies generally fall into two categories: (1) those therapies utilizing continuous daily treatment, and (2) those therapies utilizing a cyclic regimen of treatment and rest periods.
The continuous daily treatment regimens normally involve the chronic administration of relatively low doses of the bisphosphonate compound, with the objective of delivering the desired cumulative therapeutic dose over the course of the treatment period. However, continuous daily dosing has the potential disadvantage of causing adverse gastrointestinal effects due to the repetitive, continuous, and additive irritation to the gastrointestinal tract. Also, because bisphosphonates should be taken on an empty stomach followed by fasting and maintenance of an upright posture for at least 30 minutes, many patients find daily dosing to be burdensome. These factors can therefore interfere with patient compliance, and in severe cases even require cessation of treatment.
…
It is seen from current teachings that both daily and cyclic treatment regimens have shortcomings, and that there is a need for development of a dosing regimen to overcome these shortcomings.
In the present invention, it is found that the adverse gastrointestinal effects that can be associated with daily or cyclic dosing regimens can be minimized by administering the bisphosphonate at a relatively high unit dosage according to a continuous schedule having a dosing interval selected from the group consisting of once-weekly dosing, twice-weekly dosing, biweekly dosing, and twice-monthly dosing. In other words, it is found that the administration of a bisphosphonate at a high relative dosage at a low relative dosing frequency causes less adverse gastrointestinal effects, particularly esophageal effects, compared to the administration of a low relative dosage at a high relative dosing frequency. This result is surprising in view of the teachings suggesting that adverse gastrointestinal effects would be expected to increase as a function of increasing bisphosphonate dosage. Such administration methods of the present invention would be especially beneficial in treating patients that have been identified as suffering from or are susceptible to upper gastrointestinal disorders, e.g. gastrointestinal reflux disease (i.e. “GERD”), esophagitis, dyspepsia (i.e. heatburn), ulcers, and other related disorders. In such patients conventional bisphosphonate therapy could potentially exacerbate or induce such upper gastrointestinal disorders.
From a patient lifestyle standpoint, the methods of the present invention would also be more convenient than daily or cyclic dosing regimens. Patients would be subjected less frequently to the inconvenience of having to take the drug on an empty stomach and having to fast for at least 30 minutes after dosing. Also, patients would not need to keep track of a complex dosing regimen. The methods of the present invention are likely to have the advantage of promoting better patient compliance, which in turn can translate into better therapeutic efficacy.’
4 Merck does not defend the claims of the patent as granted but proposes amendments which involve the redrafting of certain of the granted claims so that the form of claims finally to be pursued are as follows:
‘1. A method of preventing osteoporosis in a human, comprising orally administering to said human a pharmaceutically effective amount comprising about 35mg of alendronate monosodium trihydrate on an alendronic acid active basis as a unit dosage according to a continuous schedule having a dosage interval which is once weekly.
2. A method of treating osteoporosis in a human, comprising orally administering to said human a pharmaceutically effective amount comprising about 70mg of alendronate monosodium trihydrate on an alendronic acid active basis as a unit dosage according to a continuous schedule having a dosage interval which is once weekly.
3. A method for treating or preventing osteoporosis in a human, said method comprising orally administering to said human a pharmaceutically effective amount of a pharmaceutically acceptable salt of alendronate, said pharmaceutically acceptable salt being selected from the group consisting of sodium, potassium, calcium, magnesium and ammonium salts, as a unit dosage according to a continuous schedule having a dosage interval which is once weekly.
4. A method for treating or preventing osteoporosis in a human, comprising orally administering to said human a pharmaceutically effective amount of risedronate, pharmaceutically acceptable salts or esters thereof and mixtures thereof, as a unit dosage according to a continuous schedule having a dosage interval which is once weekly.
5. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 35mg on an alendronic acid active basis of a bisphosphonate selected from the group consisting of alendronate, pharmaceutically acceptable salts or esters thereof, and mixtures thereof wherein said pharmaceutical composition is adapted for oral administration as a unit dosage according to a continuous schedule having a periodicity of about once-weekly.
6. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 35mg on an alendronic acid active basis of a bisphosphonate selected from the group consisting of alendronate, alendronate monosodium trihydrate or esters thereof, and mixtures thereof wherein said pharmaceutical composition is adapted for oral administration as a unit dosage according to a continuous schedule having a periodicity of about once-weekly.
7. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 35mg on an acid active basis of a bisphosphonate selected from the group consisting of risedronate, pharmaceutically acceptable salts or esters thereof, and mixtures thereof wherein said pharmaceutical composition is adapted for oral administration as a unit dosage according to a continuous schedule having a periodicity of about once-weekly.
8. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 35mg on an acid active basis of a bisphosphonate selected from the group consisting of risedronate, risedronate monosodium hemi-pentahydrate, or esters thereof, and mixtures thereof wherein said pharmaceutical composition is adapted for oral administration as a unit dosage according to a continuous schedule having a periodicity of about once-weekly.
9. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 70mg, on an alendronic acid basis, of a bisphosphonate selected from the group consisting of alendronate, pharmaceutically acceptable salts or esters thereof and mixtures thereof.
10. An oral pharmaceutical composition comprising a pharmaceutically acceptable carrier in association with about 70mg, on an alendronic acid basis, of a bisphosphonate selected from the group consisting of alendronate, alendronate monosodium trihydrate or esters thereof and mixtures thereof.’
It is convenient to proceed as if the amendments had been made. Arrow does not attack claims 4, 7 and 8 as it has no commercial interest in doing so.
Prior Patent History
Base Patent – 1983
5 The ‘base’ patent application which discloses alendronic acid, compositions containing the same and a method of inhibiting bone resorption by administering the same, was filed by Gentili SpA in 1983 claiming priority back to 1982. It was also filed in the following countries: Great Britain (GB); United States of America (US); Belgium; France; Germany; Italy; Netherlands; Luxembourg; Sweden; Japan; Switzerland.
6 The patents granted in each of these countries contain different claims. For example the GB patent 2118042 contains claims directed specifically to alendronic acid and pharmaceutical compositions containing alendronic acid or salts thereof, whereas the corresponding US patent US4721077 contains a single claim directed to the method of inhibiting bone resorption by administering an effective amount of alendronic acid to a patient in need thereof. This patent has no Australian counterpart. Both the above GB and US patents have been the subject of revocation proceedings. The US patent has been upheld as valid although the GB patent has been revoked.
7 The United Kingdom specification included the following:
‘The pharmaceutical compositions according to the present invention may be prepared for use in the form of capsules or tablets or in solution for oral administration or for systemic use. The compositions are advantageously prepared together with inert carriers such as sugars (saccharose, glucose, lactose), starch and derivatives, cellulose and derivatives, gums, fatty acids and their salts, polyalcohols, talc, aromatic esters.
The pharmaceutical compositions can be administered by the oral route at doses from 25 to 3200 mg/die or by the parenteral route at doses from 15 to 300 mg/die. Treatment is carried out for 7 days or for 3 months’ courses, repeated according to needs.’
8 Claims 2 and 3 of the GB patent were:
‘2. A pharmaceutical composition according to claim 1 in a form for oral administration.
3. A pharmaceutical composition according to claim 1 in a form for systemic administration.’
The Trihydrate Salt Patent – 1990
9 In 1990 Merck filed a patent application claiming priority back to 1989 that was granted as Australian Patent 625704 which is currently in force and is due to expire on 8 June 2010. That Australian patent includes claims directed to the specific alendronate monosodium trihydrate species, compositions containing the same, a process for preparing alendronic acid or salts and methods for treating or preventing osteoporosis by administering the same.
10 The summary of the invention in the specification included:
‘According to yet a further embodiment of this invention there is provided a pharmaceutical composition comprising a therapeutically effective amount of a compound of the foregoing embodiments together with a pharmaceutically acceptable carrier, diluent and/or excipient.
According to yet a further embodiment of this invention there is provided a method of treating or preventing a disease of bone resorption comprising administering to a person in need of such treatment a therapeutically effective amount of the compounds or compositions of the foregoing embodiments.’
11 The claims included:
‘20. A method of treating or preventing a disease of bone resorption comprising administering to a person in need of such treatment an effective amount of the compound of any one of claims 14, 15, 16, or 17, or the composition of claim 18 or 19.
21. The method of claim 20, wherein said disease is hypercalcemia of malignancy, Paget’s disease or osteoporosis.
22. The method of claim 20, wherein said disease is osteoporosis.
…
25. A method of treating or preventing a disease of bone resorption comprising administering to a person in need of such treatment an effective amount of the compound of claim 23, or the composition of claim 24.
26. The method of claim 25, wherein said disease is hypercalcemia of malignancy, Paget’s disease or osteoporosis.
27. The method of claim 25, wherein said disease is osteoporosis.’
Narrative of Facts
12 The narrative of the facts which follows is largely drawn from the chronologies prepared by the parties and much of it is not controversial. Some of it is reflected in the extracts from the specification of the patent in suit already set out.
13 Bisphosphonates have been known since the 1970s to be inhibitors of bone resorption. The clinical application of bisphosphonates including their use, effectiveness and side effects in the treatment and prevention of osteoporosis was well known. Gastrointestinal (GI) side effects were well known. Etidronate and clodronate were each bisphosphonates developed for clinical use. Pamidronate followed. It was the first aminobisphosphonate compound developed for clinical use in the treatment of osteoporosis. Clinical trials were organised by Ciba-Geigy in 1989–1990. The trials of oral administration of pamidronate were discontinued because of GI side effects. The trial experience was published in November 1994 in Vol 4 of the periodical Osteoporosis International in an article by Lufkin et al entitled ‘Pamidronate: An Unrecognised Problem in Gastrointestinal Tolerability’. The 1995 edition of Bisphosphonates in Bone Disease by Professor Fleisch reported (at p 152) that:
‘Oral pamidronate as well as other aminobisphosphonates can give rise to dose-related gastrointestinal disturbances that may be severe’.
That report related to oral doses of 300 mg or more daily.
14 In about 1987 or 1988, Merck acquired the rights to the base Gentili patents for alendronate and set about exploiting them. It was necessary to satisfy the Food and Drug Administration (FDA) of the United States of efficacy and safety pursuant to the US regulatory regime concerning drugs for the treatment of humans. Phase II clinical trials commenced in 1991 with the Harris study of six weeks on 65 healthy post-menopausal women. It was followed by the two year Chesnut study in 1992–1993 as to 5, 10, 20, and 40 mg orally daily on 188 post-menopausal women with osteoporosis. That study was reported in September 1995 in the periodical American Journal of Medicine, Vol 99 in an article by Chesnut et al ‘Alendronate Treatment of the Postmenopausal Osteoporotic Woman: Effect of Multiple Dosages on Bone Mass and Bone Remodeling’ (the Chesnut article).
15 The Phase III clinical trials were conducted over three years between 1993 and 1995 in the so-called Liberman study. That was an international trial on 994 women with post-menopausal osteoporosis treated with 5 or 10 mg daily for three years or 20 mg daily for two years followed by 5 mg daily for one year. The results of that trial were reported in December 1995 in the periodical New England Journal of Medicine, Vol 333, No 22 in an article by Liberman et al entitled ‘Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis’ (the Liberman article).
16 Lunar Corporation was a US company with a business that specialised in bone densitometry. The principal was Dr Mazess (whose qualifications and experience were in the fields of radiology and medical physics). It did business with Merck from time to time. Several times a year it published a magazine entitled ‘Lunar News’ which contained articles on topics of interest related to its business. The articles footnoted relevant journal references. It will be necessary to examine the question of the publication of Lunar News in more detail later. The December 1995 issue included an article headed ‘Update: Bisphosphonates’ and commenced as follows:
‘Bisphosphonates are the preferred agents among researchers today for treating osteoporosis, although they are not yet part of routine clinical management. This will change in the near future because alendronate (Fosamax by Merck) gained FDA approval in the U.S. in October 1995.’
(original emphasis)
The article proceeded to review developments in relation to alendronate and other bisphosphonates including risedronate. The Chesnut article was footnoted amongst others.
17 By October 1995 alendronate was approved for sale and was on sale by Merck in the United States under the trade name ‘Fosamax’ on the basis of a dosage of 10 mg per day for the treatment of osteoporosis and 40 mg per day for the treatment of Paget’s disease.
18 In March 1996 the Clinical Development Oversight Committee (CDOC) of Merck considered adverse GI events (AE) which had occurred with the use of Foxamax after marketing commenced. One of the consultants involved was Dr Piet de Groen from the Mayo Clinic. The observations were summarised as follows:
‘… that the incidence and severity of esophageal AEs appear to be greater than observed in clinical trials, that improper dosing is likely to be common in the marketed use of alendronate (especially in patents with post-marketed reports of esophageal AEs), and that only rare cases (n=6) exist of severe esophageal AEs in patients who appear to have taken alendronate correctly. The Team concluded that alendronate appears to cause chemical esophagitis in a small number of patients, which may be severe, improper dosing may be a factor, and the role of concomitant therapies and prior UGI disorders are not established with existing data.’
19 It was agreed that action should be taken and the assignments included the following:
‘FOSAMAX – Project Team/Pharm R&D – Discuss the possibility of new formulations of alendronate which may alleviate some of the dosing difficulties.’
20 On 15 March 1996 a letter was sent to physicians (known internally as the ‘Dear Doctor letter’) as follows:
‘We would like to call your attention to esophageal side effects in patients taking FOSAMAX Since market introduction of the product, some of these side effects have been of greater severity than we observed in our controlled clinical trials. It is clear from the case reports received by Merck & Co., Inc. that FOSAMAX can be irritating to the esophagus but that careful adherence to the dosing instructions can reduce the potential for these side effects. In a large proportion of cases reported, there is evidence that FOSAMAX was not dosed in the manner recommended in the Product Circular. We have therefore made modifications to the Product Circular (and Patient Package Insert) that we believe will further reduce the risk of adverse reactions such as esophagitis, esophageal erosion or ulceration. Merck & Co., Inc. is writing to you directly as the most rapid way to bring this important information to your attention.
FOSAMAX is an aminobisphosphonate, a selective inhibitor of bone resorption that is the first nonhormonal therapy indicated for the treatment of osteoporosis in postmenopausal women (the daily oral dosage is 10 mg). FOSAMAX is also indicated for the treatment of Paget’s disease of bone (the daily oral dosage is 40 mg for six months). In controlled clinical trials of three years’ duration in postmenopausal women with osteoporosis, FOSAMAX was generally well tolerated. Like other bisphosphonates, however, FOSAMAX was recognized to have the potential to cause local irritation of the upper gastrointestinal mucosa. In ongoing, carefully-monitored studies of over 12,000 patients, esophageal irritation has been no different in incidence or severity than observed in the completed three-year trials. (As you know, esophageal ulcers, considered to be drug related, were reported in 1.5% of the patients taking FOSAMAX 10 mg in three-year trials.)
As part of our customary postmarketing surveillance, we have become aware of a number of case reports of esophagitis and esophageal ulceration in which patients have presented with difficulty or pain on swallowing, and/or retrosternal pain. When endoscopy has been performed, the findings have been consistent with a chemical irritation of the esophagus.
Although infrequent, the reports of esophagitis/ulceration have generally been of a more severe nature than observed in either previous clinical trials, or in ongoing studies of FOSAMAX (alendronate sodium tablets). In a large majority of the postmarketing reports, it appears patients were not compliant with our recommended dosing instructions (for example, patients were taking FOSAMAX with little or no water, taking it at bedtime and/or lying down within minutes after taking it, etc.). Several patients continued taking FOSAMAX after the occurrence of symptoms suggestive of esophageal irritation. In a few cases, patients have been found to have previously undiagnosed esophageal disorders.
We therefore strongly advise strict compliance with the dosing instructions, which have been updated in the Product Circular, and are summarized in the attachment. Please read them carefully, and in the case of any question contact the Merck National Service Center at (800) 672-6372. Enclosed is a copy of the revised Product Circular. Merck has been taking, and is continuing to take, a number of steps to ensure that physicians, pharmacists, and patients understand the importance of proper dosing of FOSAMAX. We believe that the risk of esophageal irritation can be substantially decreased with adherence to these recommendations.
At Merck, our primary concern remains the safety and well-being of the patients who use our products. We appreciate the continuing efforts of doctors, pharmacists and other healthcare professionals in communicating dosing information to patients. We truly value your trust in us and thank you very much for your time and attention to this matter.’
21 A comprehensive report followed. That report included the following:
‘II. PRECLINICAL DEVELOPMENT OF ALENDRONATE
Alendronate was licensed by Merck & Co., Inc. from Gentili S.p.A. for treatment of disorders associated with increased bone resorption. The formulation that had been developed by Gentili was the free acid, which had low solubility and had been shown to be associated with esophageal ulceration in two patients treated with an oral formulation. In recognition of the potential for mucosal irritation of this formulation, alendronate was reformulated by Merck as the sodium salt. Alendronate sodium has a more neutral pH and greatly improved solubility characteristics relative to the original acid formulation, and was very well tolerated in animal studies. Gastric ulceration was observed in rats treated with alendronate at a dose of 10 mg/kg (equivalent to approximately 50 times the human osteoporosis treatment dose of 10 mg per patient per day). Ulceration was not observed at 5 mg/kg, although some gaseous distention was seen at that dose, presumably due to effects on the GI flora. Furthermore, no GI ulceration was observed in dogs treated with doses of up to 8 mg/kg/day over one year. Both in the rat and dog studies, alendronate was given by gavage, and therefore exposure of the esophagus to alendronate was limited to reflux that occurred upon removal of the gavage tube.
Based upon the experience in preclinical studies, it was considered that the alendronate sodium formulation would be acceptable for clinical use, although irritation of the upper gastrointestinal mucosa was clearly considered to be a potential side effect of alendronate treatment. For this reason, the dosing instructions utilized in the clinical trials were developed to reduce the potential for sticking of the alendronate tablets within the esophagus. Specifically, patients were instructed to take alendronate with a full glass (six to eight ounces) of water and to take study medication while upright and not to lie down for at least 30 minutes thereafter. Several published studies identify upright position and volume of water as critical for ensuring rapid delivery of tablets to the stomach. The amount of water most commonly referred to as adequate for this purpose is at least 100 mL, which is approximately 4 oz (Channer and Virjee, 1986; Hey et al., 1982).’
22 The report summarised the clinical trial experience as follows:
‘… this review of all studies included in the current osteoporosis treatment database has found no evidence of increase in either all, serious, or clinically severe esophageal adverse reactions with alendronate 10mg. There may be a slight increase in such events at the 20 or 40mg dose. The labeling adopted in the prescribing information therefore remains an accurate description of the clinical trial experience with alendronate.’
(emphasis added)
23 Another relevant part of the report was:
‘IV. PAGET’S DISEASE OF BONE
The studies in Paget’s Disease of bone (N = 235) include four protocols with either 3 or 6 months of alendronate therapy at higher doses (20, 40 and 80 mg) of alendronate. As in the osteoporosis treatment studies, patient with active upper GI disease or NSAID use were prohibited from entering these studies. There was only one serious and/or clinically severe case of esophagitis reported (Study 059001 AN 1226). This patient is interesting in that she initially would lie down after taking alendronate, but once her improper dosing was corrected, the esophagitis cleared, and she was able to go back onstudy therapy (alendronate 40 mg) without further adverse experiences. A table of the incidence of all upper GI AEs in the studies of Paget’s disease of bone is provided in Appendix 3.’
(emphasis added)
Interestingly, the experience in a Fracture Intervention Trial (ie prevention) at 5 mg of alendronate or placebo, with the dose for the active treatment group increased to 10 mg at month 24, was consistent with the osteoporosis treatment clinical studies at the higher dose in relation to GI effects ie no dose response was observed.
24 The conclusion drawn in the report from post marketing experience was:
‘Therefore, in the majority of reported cases where sufficient information could be obtained, it appears that patients who developed clinically severe or serious esophageal AEs either took FOSAMAX with too little water and/or lay down after dosing and before their first food of the day and/or continued to take FOSAMAX after their onset of esophageal symptoms. In view of these clinical findings, it is very important to ensure that full and adequate dosing instructions are provided to all patients who are prescribed FOSAMAX, and that patients who might be at increased risk for adverse esophageal reactions be clearly specified in the label.’
The summary concluded as follows:
‘Given the very widespread marketed use of FOSAMAX as well as the known potential for mucosal irritation, the postmarketing experience is not surprising, and the number of reports of upper GI AEs appears consistent with expectations based upon the clinical trials experience. However, the incidence of severe esophageal AEs can clearly be reduced by concerted actions to maximize the potential for patients to adhere to the dosing instructions.’
(emphasis added)
25 The April 1996 issue of Lunar News included the following:
‘One of the difficulties with alendronate is its low oral bioavailability. When taken with water in a fasting state, only about 0.8% of the oral dose is bioavailable [Gertz BJ, Holland SD, Kline WF, Matuszewski BK, Freeman A, Quan H, Lasseter KC, Mucklow JC, Porras AG, (1995) Studies of the oral bioavailability of alendronate. Clin Pharmacol Ther 58:288–298]. Even coffee or juice reduces this by 60%, and a meal reduces it by >85%. Alendronate must be taken, after an overnight fast, 30–60 minutes before breakfast. Subjects should remain seated or standing; a very small group of patients have reported some upper gastrointestinal distress if this is not done. This regime may be difficult for the elderly to maintain chronically. An intermittent treatment program (for example, once per week, or one week every three months), with higher oral dosing, needs to be tested. A sustained response has been demonstrated to intravenous administration of high-dose alendronate [Vasikaran SD, Khan S, McCloskey EV, Kanis JA (1995) Sustained response to intravenous alendronate in post-menopausal osteoporosis. Bone 17:517–520].’
(emphasis added)
Several sources are footnoted, including Chesnut and Liberman. Lunar Corporation had a website operating by then.
26 A meeting of the Research Management Committee (RMC) of Merck on 11 April 1996 decided that the Pharmaceutical Research & Development team should undertake studies on buffered formulations of the Fosamax tablet, ie formulations to reduce acidity.
27 Between March and May of 1996 Dr Peter (Peter) of Merck conducted esophageal irritation studies on dogs. The first gastric reflux study was performed between 21 March and 19 April. The second gastric reflux study was performed between 2 May and 10 May. The third gastric reflux study was performed between 20 May and 24 May. A single study was performed between 25 April and 2 May. The first test included a single infusion of alendronate at 0.2 mg/ml in simulated gastric juice at pH 2 with no esophageal irritation observed. Dr Yates (Yates) and Dr Santora (Santora) each assisted in the design of the studies and were familiar with the results. Each was a named inventor in the patent in suit. Yates was, at that time, Senior Director, Clinical Research Endocrine/Metabolic and Santora was a colleague of his. Yates says that he concluded from those trials that it might be possible to administer a high dose of alendronate as a single dose and that, even if alendronate were to reflux into the esophagus, the potential for esophageal damage would be low whilst, in contrast, repeated daily exposure to alendronate resulted in significant esophageal injury similar to that seen in human case reports. That is claimed to be the invention. That claim is challenged by Arrow. As will be seen, I reject it.
28 On 12 June the RMC received a report on the dog studies. It was decided that the anesthetized dog model was very predictive in the evaluation of esophageal irritation with alendronate sodium. Additional studies to be performed were described. It is noteworthy that these did not include any single dose study. It was also reported that seven formulations were undergoing pharmaceutical evaluation as alternatives to the marketed oral tablet (10 mg). The RMC supported the continuation of that program. There was no mention of alternative dosages.
29 Fosamax was initially registered in Australia pursuant to the Therapeutic Goods Act 1989 (Cth) in July 1996 at a dosage of 10 mg daily (for treatment of osteoporosis) and 40 mg daily (for treatment of Paget’s disease).
30 The July 1996 issue of Lunar News included the following as part of an article headed ‘Update: Bisphosphonate’:
‘Some U.S. physicians are reluctant to treat because of: (a) side effects, (b) difficulty of dosing, and (c) high costs ($700/year). First, Merck recently sent a letter to physicians warning of esophagitis. Some physicians report that 5 to 15% of patients experience gastric and/or esophogeal distress, but most have seen no side effects. Serious side effects of ulceration and stricture appear rare [Maconi G, Porro GB (1995) Multiple ulcerative esophagitis caused by alendronate. Am J Gastroenterol 90:1889–1890]. Second, some patients also stop alendronate because of the dosing difficulty. The limited bioavailability of alendronate (0.8%) requires that it be taken on an empty stomach upon awakening with a full glass of water (not tea, coffee, or juice), and the patient must remain upright for 30 to 60 minutes [Gertz BJ, Holland SD, Kline WF, Matuszewski BK, Freeman A, Quan H, Lasseter KC, Mucklow JC, Porras AG (1995) Studies of the oral bioavailability of alendronate. Clin Pharmacol Ther 58:288–298]. A few elderly women can tolerate this regime for a only [sic] week or two. Third, some U.S. patients, whose insurance does not cover drug costs, are finding alendronate expensive. Some health organizations are even recommending use of lower-cost cyclical etidronate. Alendronate is the first satisfactory and effective therapy for the 85% of patients who do continue. Merck is attempting to reach an even larger group of women by promoting densitometry of the hand and forearm. This promotion of outmoded densitometry has antagonized leading physicians (who use axial densitometry). This antagonism possibly could be a factor in the unexpectedly high frequency of side effects and dosing difficulties. The use of forearm BMD in relation to bisphosphonates seems surprising, since the forearm shows no increase with treatment.
The difficulties with oral bisphosphonates may favour their episodic (once/week), or cyclical (one week each month) administration. Even oral alendronate potentially could be given in a 40 or 80 mg dose once/week to avoid dosing problems and reduce costs.’
(emphasis added)
31 An exchange of emails between Merck personnel in early August 1996, that assumes some importance in light of later events, was as follows:
‘From: Drake, Paul J.
To: Santora, Arthur C.; Yates, A. John
Subject: 40 mg once per week
Date: Friday, August 02, 1996 4:29PM
Art,
As you may recall, Per Wold-Olson brought up the issue of some Italian investigators looking into dosing FOSAMAX at 40mg once per week to get around the daily restrictions and potential for AE’s with the 10mg dose. While this may sound far fetched, I think we will need to respond to this issue with our position on this proposed regimen. Can you help me out with this?
Paul’
‘Relevant recent communication:
From John Yates to Paul Drake:
Paul
This issue was discussed at the CT meeting yesterday.
40 mg/week is approximately equal to 5 mg/day, so 80 mg would be the more appropriate equivalent treatment dose.
Issues are:
1) Possible increased GI toxicity because of high dose. However, this is speculative because single dosing in dogs was not a problem, so perhaps the mucosa can rebound if given 6 days without alendronate. This would need to be tested in the dog model.
2) Compliance might be worse because of the lack of routine. However, this is uncertain, because motivated patients might much prefer foregoing breakfast once a week rather than every day.
3) Conceivable, efficacy of one weekly dosing could be less than daily, although based upon the animal studies this seems unlikely.
Whether we like it or not, Per has a point, and this could be considered for further development if there is Marketing interest. We would need an SOI (groan!)
John’
‘From: Santora, Arthur C.
To: Melton, Mary; Eng, Marvin F.; PRTM; Jackson, Donna K.; Santora, Arthur C.; Yates, A. John; Johnson, Michele A.; Katdare, Ashok V.; Radican, Larry; Drake, Paul J.; Whiteford, Donna
Cc: DiCesare, Elizabeth A.
Subject: Once a week Fosamax in Prevention of Osteoporosis
Date: Sunday, August 04, 1996 9:53 AM
Folks,
Per Wold-Olson recently raised the issue of a weekly dose of FOSAMAX (see communication below) as a way of producing a more convenient dosing schedule. For a variety of reasons, weekly dosing in an elderly osteoporotic population is not a great idea. The dose would have to be at least 80 mg almost certainly leading to an increased incidence of pill esophagitis, and elderly individuals – frequently on other daily (or several times a day) medications – are very likely to be confused by an unusual weekly dosing schedule.
However, younger recently menopausal women are a different story. In an informal poll (admittedly “non-scientific”) of a few women between 35 and 60 years, the overwhelming preference was for a weekly dose, even after considering that the possibility of gi side effects may be greater per dose. In fact, I haven’t found anyone who preferred a daily dose for her own use. The BMD and biochemical effect of a 40 mg per week dose is likely to be indistinguishable from 5 mg daily in a prevention population. Also, in a younger population where esophageal motility problems and gastroesophageal reflux are less common, the gi side effect profile of 40 mg weekly will probably be indistinguishable from placebo (or 5 mg daily).
Anyway, with all the hype about raloxifene and droloxifene in prevention, we need an advantage. In this population, a once a week dose of FOSAMAX would provide that advantage. It may be a little late to include this issue in the Stage 0 review at the Tactical PAC on August 19th, but if I were to prioritize the alternate formulations and dosage forms development with a 40 mg weekly dose on the list, the 40 mg weekly dose would be number 1. I realize that there may be pricing implications which I haven’t considered, but keep in mind that Merck will make no money on a prescription for raloxifene.
Art’
(emphasis added)
32 On 3 September 1996 a Tom Pyron from Merck wrote a memorandum to a Rick Roberts of Merck concerning the July issue of Lunar News. The text was:
‘I recently received LUNAR® News, the infomag written by the folks at LUNAR Corp., Madison, WI. There are a number of articles of interest, and many comments that MRL might want to comment on at ASBMR, as Dick Mazess I’m sure will be in attendance.
Attached are copies of some of the leading articles in News, in which unflattering comments are made about peripheral BMD measurements (page 6), pQCT (page 10), biochemical markers (page 11), the Hologic QDR reference values (page 14), and alendronate (page 23), while estrogen received some very positive commentary (page 20).’
(emphasis added)
The memorandum from Pyron was widely disseminated. The recipients included Yates.
33 The 18th annual meeting of the American Society of Bone and Mineral Research (ASBMR) was held on 10 September 1996 at Seattle, Washington. The agenda included the following topics:
· ‘The Clinical Profiles of Bisphosphonates
· New Data on the GI Effects of Bisphosphonates
· A Real World Look at a Bisphosphonate’s Efficacy, Safety and Tolerability: The UK GPRD Database.’
Both Yates and Mazess attended the meeting but there is no satisfactory evidence of what, if anything, occurred between them.
34 On 1 October 1996 Fosamax was released for sale in Australia for treatment of osteoporosis at 10 mg and Paget’s disease at 40 mg per day.
35 In October 1996 the New England Journal of Medicine Vol 335 No 14 contained articles and items concerning esophagitis and alendronate including P C De Groen et al, ‘Esophagitis associated with the use of Alendronate’, D O Castell, ‘Pill Esophagitis – The case of alendronate’ and V A Liberman and L J Hirsch, ‘Esophagitis and Alendronate’.
36 On 1 November 1996 Fosamax received Pharmaceutical Benefits Scheme (PBS) listing in Australia for treatment of Paget’s disease at 40 mg daily. This involved a substantial government subsidy of the cost of the drug to the patient.
37 The December 1996 edition of Lunar News included the following under the heading ‘Update: Bisphosphonate’:
‘At the American Society of Gastroenterology meeting this October in Seattle, clinical investigators reported that alendronate, like aspirin, could be a potent irritant to the esophagus and gastrointestinal tract. The influence, if any, on Helicobacter pylori infestation, ulcers, and adenocarcinoma is unknown.
At recent meetings, like the American Society for Bone and Mineral Research and the American College of Rheumatology, many U.S. and European prescribers informally discussed the incidence of alendronate side effects (10% to 20%) which were higher than those expected. One explanation is that side-effects from any drug are common in the elderly, and another explanation is that physicians may be treating patients with a history of reflux or gastritis. A report published in the New England Journal of Medicine [de Groen PC, Lubbe DF, Hirsch LJ, Daifotis A, Stephenson W, Freedholm D, Pryor-Tillotson S, Seleznick MJ, Pinkas H, Wang KK (1996) Esophagitis associated with the use of alendronate. N Engl J Med 335:1016–1021.] attracted attention to the problem, and suggested that half or more of the gastroesophogeal problem could be minimized by proper administration [Castell DO (1996) “Pill esophagitis” – the case of alendronate. N Engl J Med 335;1058–1059.]. Alendronate must be taken upon awakening with a full glass of water, and the patient cannot recline. While some physicians claimed the side effects were not due to poor administration, the number of reported adverse incidents per patient has dropped since Merck sent out a letter to physicians. A total of 200 incidents had been reported to the FDA as of September 1996, of which only 20% were serious. There may be some underreporting of mild incidents because affected patients terminate treatment, but not of serious side-effects, which are rare. Experts still view alendronate positively, but etidronate may be preferred in the most elderly patients since its side effects are lower. All bisphosphonates can cause stomach upset, cramps, and diarrhea, but amino-bisphosphonates (pamidronate and alendronate) have a higher potential for inducing esophagitis and ulceration.’
And later:
‘Clinicians in the U.K., Canada, Australia and Sweden, where there has been extensive experience with etidronate seem to prefer this agent because of its lower cost, (one-third to one-fourth that of alendronate), and lower incidence of esophageal and gastrointestinal side effects.’
38 On 9 January 1997 John Gurney, a Merck Marketing Associate, wrote to various Merck personnel, including Yates and Santora, regarding unfavourable articles in the December 1996 edition of Lunar News and attaching copies.
39 On 1 February 1997 Fosamax received PBS listing in Australia for the treatment of osteoporosis at 10 mg daily.
40 By letter of 7 February 1997 Mazess wrote to Mr Ray Gilmartin (Gilmartin), the Chief Executive Officer and President of Merck, concerning, among other things, what he described as the dramatically deteriorating relations between Lunar and the Bone Measurement Institute (BMI) of Merck, particularly because of the attitude of Mr Jeremy Allen (Allen), the Vice President of the Osteoporosis Therapeutic Business Group and Director of the BMI. The reply to that letter was from Mr David Anstice, President, Human Health – The Americas of Merck dated 1 April 1997. It proposed a meeting with Dr Lou Sherwood (Sherwood), (Senior Vice President, Medical & Scientific Affairs, Merck US Human Health and President, BMI), Allen and Yates at Lunar’s office at Madison, Wisconsin. That meeting was arranged for Wednesday, 21 May at 8.30 am.
41 The April 1997 edition of Lunar News contained an article entitled ‘Update: Bisphosphonate’. Amongst other things, it claimed a very high effective cost for preventive treatment by alendronate. It contained the following passage:
‘One way to reduce the relatively high cost, and the potential side-effects, of potent oral bisphosphonates may be to dose only two or three times per week rather than daily. In pigs, the bisphosphonate dose can be reduced without adverse effect on skeletal response by giving a standard “daily dose” every fourth day, or for 5 days out of 20 [de Vernejoul MC, Pointillart A, Bergot C, Bielakoff J, Morieux C, Laval Jeantet AM, Miravet L (1987) Different schedules of administration of (3 amino-1-hydroxypropylidene)-1,1 bisphosphonate induce different changes in pig bone remodelling. Calcif Tissue Int 40:160–165]. In the human, there is little difference between 5 and 10 mg/day of alendronate, so the 10 mg dose theoretically could be given only 3X a week or the 40 mg dose once per week. Serum biochemical markers could be used to titrate dose downward. If turnover remained low after three months at 3X/week, the dose could be reduced to 2X/week or lower. These new clinical approaches to dosing need to be tested in at least short-term trials.
An alternative approach is to give high doses of a potent bisphosphonate once or twice a year. Vasikaran et al [Vasikaran SD, Khan S, McCloskey EV, Kanis JA (1995) Sustained response to intravenous alendronate in post-menopausal osteoporosis. Bone 17:517–520] found that alendronate infusion over four days produced a decrease of bone turnover for 6-months, and a 3% increase of spine BMD. The dose received (30 mg) would be equivalent to that provided by one year of oral treatment at 10 mg/day considering <1% absorption for oral treatment. Theoretically a small dose (7.5 mg) of alendronate could be infused once every 3 months. This, in fact, is the preferred administration route for ibandronate. These alternatives to oral treatment eliminate (a) the inconvenience of daily oral administration, (b) non-uniform effects due to variations in absorption, and (c) gastro-esophageal side effects. Finnish investigators [Heikkinen JE, Selander KS, Laitinen K, Arnala I, Vaananen HK (1997) Short-term intravenous bisphosphonates in prevention of postmenopausal bone loss. J Bone Miner Res 12:103–110]found that high-dose intravenous etidronate or clodronate (900 mg given on 3 occasions during one month) maintained spine and femur BMD over the successive two years. These bisphosphonates produce even fewer flu-like symptoms than infused amino-bisphosphonates and thus may prove even more acceptable for “minimally invasive” long-term treatment.’
(emphasis added)
42 On 7 May 1997 Santora sent the following email to Peter:
‘Peter,
You have probably heard that we are considering use of a 40 mg dose of FOSAMAX once a week for the prevention of osteoporosis (expected efficacy similar to 5 mg daily). We are less seriously considering an 80 mg dose once a week for treatment and/or 40 mg twice a week for treatment. John Yates mentioned that a conclusion from the dog esophageal irritation studies you conducted was that esophageal exposure to acidic solutions of alendronate had to be carried out for several days before substantial injury was apparent. The hypothesis was raised that a relatively high dose might be well tolerated, even in a patient with esophageal reflux, if it were administered weekly, with time between doses for recovery from any damage which might occur with any one dose.
First, do you think this hypothesis is viable? Second, could the dog esophageal model be used to test the hypothesis?
Art’
(emphasis added)
43 On 12 May 1997 a Merck Technical Review Meeting concerning Fosamax was held. The participants included Yates, Dr Anastasia Daifotis (Daifotis), who is another alleged inventor, and Santora. It considered a paper prepared for the occasion. The introduction to the paper included the following:
‘Another area of continued investigation is the development of alternative formulations and alternative approaches to dosing that may make FOSAMAX easier to take and/or safer than the current regimen. The relative clinical importance of the various clinical initiatives, their likelihood for success and timing will be discussed. Also, the proposed study designs for the repeat bioavailability study with the oral buffered solution and the pilot beverage interaction study will be reviewed. Since much of the drive to develop these new approaches is based upon Marketing needs, the Marketing group will briefly present their perspectives on the potential value of the alternative formulations, tablet image and the needs for increased dosing flexibility.’
(emphasis added)
44 One segment of the paper was on an alendronate alternate formulation development program, which was said to have been initiated to address marketing needs and address the new findings from the safety studies conducted in an isolated dog esophagus model. Less frequent administration of alendronate at a higher dosage rate is not a formulation but, rather, a dosage regime.
45 On 20 May 1997 a Tactical Review of the Fosamax Stage 1 Product Development Plan by the Tactical Product Approval Committee (TPAC) took place. That meeting is most significant as it is the first official recognition within the decision making processes of Merck of the possibility of an alternative dosage regime for Fosamax in relation to the treatment of osteoporosis. For that reason, and in the light of the issues in the case, I shall set out significant parts of the report that was prepared for the meeting. Yates prepared part of the report and he and Santora elaborated upon the proposal at the meeting. An important part of the report was the analysis of competition, particularly future competition, with concentration upon Proctor and Gamble with risedronate and Eli Lilly with raloxifene. Another was the development of further applications for Fosamax. Another was the availability of patent protection. These are illustrated by the following diagram and table which were included in the report:
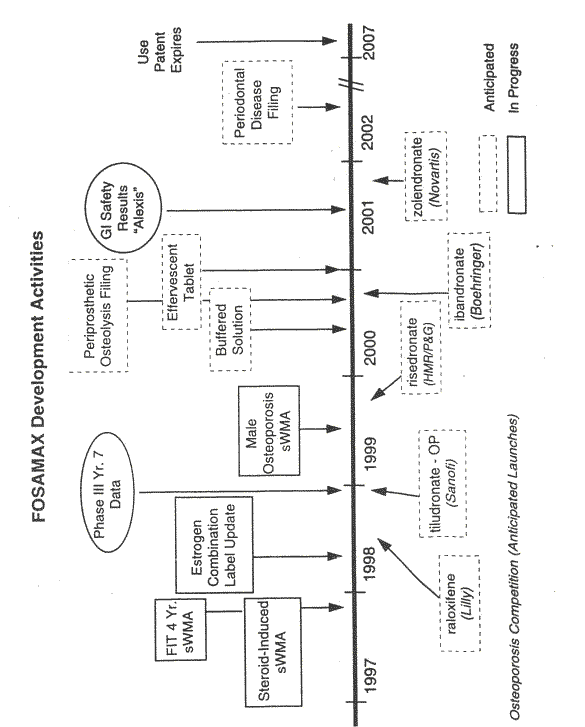
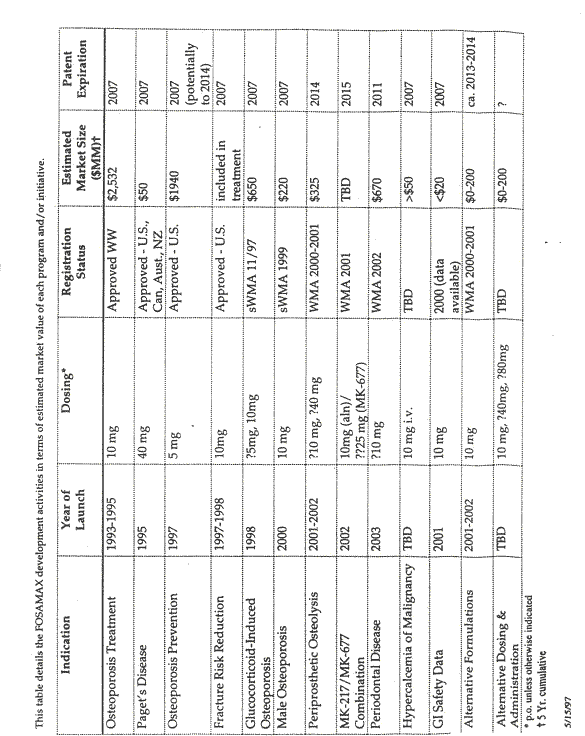
46 Key marketing needs for alternative dosing were said to include:
‘1. Development of alternative dosage forms/formulations that eliminate the food, beverage and posture contraindications or at least make it easier and more convenient for general osteoporosis patients to comply with the current recommendations.
2. Development of alternative dosage forms or product images that either reduce or have a perceived ability to reduce the potential for gastrointestinal irritation relative to the current oral tablet.’
It was said that:
‘Alternative timing of dosing is being considered. From a scientific perspective, once weekly dosing with a dose equivalent to the cumulative weekly dose is likely to be effective, and may prove to be as safe, possibly safer, than daily dosing. Patient inconvenience would clearly be minimized. However, such an approach has significant pricing considerations that need to be addressed.’
(emphasis added)
47 That part of the report which considered alternative dosing regimes in detail was as follows:
‘C. Alternative Dosing Regimens
To overcome the perception of rigorous dosing and administration schedules, in addition to alternative formulations, alternative dosing of the tablet form should be considered. Once weekly oral dosing with the same total weekly dose could be offered as an alternative to daily dosing. This would probably have greater patient acceptance and is unlikely to have a greater potential to induce upper GI irritation.
In order to obtain regulatory approval for this regimen one would need to show a similar effect between daily and weekly dosing of alendronate. A potential study design would involve a 6 month randomized, double-blind, placebo-controlled study with the following treatment groups:
· alendronate 5 mg daily
· alendronate 10 mg daily
· alendronate 35 or 40 mg weekly
· alendronate 70 or 80 mg weekly or 35 or 40 mg twice weekly.
The endpoints would include bone mineral density and biochemical markers both which [sic] have been repeatedly shown to demonstrate an effect with alendronate within 6 months.
A possible advantage of using the 35 or 70 mg doses is that these are the same total amounts of drug currently being used with 5 or 10 mg daily dosing. This could be important with regard to the perception of long-term bone safety with alendronate, especially since 5 year data are already currently available. Also, it may be possible to patent the weekly regimen, potentially with specific reference to the 35 and 70 mg doses. A patent would be more achievable if a rationale for considering possible improved safety could be put together based upon the dog esophageal model safety assessment testing. This is conceivable, since a single exposure, even at 7 times the concentration could potentially have less cumulative irritant effect than repeated exposure to a lower concentration. Indeed, in the alendronate dog esophageal irritation studies, single infusions of alendronate (0.1 or 0.2 mg at pH 2.0) did not induce gross or histologic injury. However, recurrent 5-day dosing did show histologic injury. Additional animal studies could be used to show that weekly dosing of a higher alendronate dose has less of a potential for esophageal injury than daily dosing. Human studies could show that alendronate daily or weekly are equally safe dosing regimens, but since esophageal adverse experiences are rare, it would require very large numbers to prove that weekly dosing is superior. If weekly dosing is patentable, this regimen would allow for extension of the FOSAMAX patent to 2018. One key concern would be a potential negative impact of once (or twice) weekly dosing on pricing.
There is human safety data available on these higher doses. The largest experience is derived from the Paget’s Disease studies including safety data on over 150 patients randomized to receive 3–6 months of daily treatment with alendronate 40 or 80 mg. This data is supplemented by short term clinical pharmacology studies with doses up to 100 mg. In all theses [sic] studies, the 40 and 80 mg doses were well tolerated even when given on a daily basis, although daily treatment with alendronate 40 mg was associated with a moderate increase in upper GI adverse events in the Phase II study of treatment of osteoporosis (Protocol 026).
The major regulatory issues include the need to provide a new CMC package including stability information and review of this information by the agencies for the new doses. Additionally there is concern that some agencies may require data beyond 6 months, although approximate equivalent efficacy could be demonstrated in 6 months’.
(emphasis added)
The summary of characteristics was as follows:
|
| ATTRIBUTES | CHARACTERISTICS AT FILING | CHARACTERISTICS AT LAUNCH | PROMOTIONAL BENEFITS/ | PATENT STATUS |
| Intermittent dosing | · 40 mg weekly for prevention · 40 mg twice a week for treatment | Same restrictions for drugs, fluids, posture and food on day of dosing only. | Equivalent efficacy to daily dossing. Pricing considerations make this less attractive | Increased dosing flexibility, convenience. | TBD |
(emphasis added)
In the section relating to required resources, the following was included:
‘Parallel activities are also on-going to support a trial to test the safety and efficacy of once-a-week therapy for prevention and twice-a-week therapy for treatment using the 40 mg dose.’
(emphasis added)
48 The proposal was accepted by the TPAC at the meeting of 20 May 1997. An immediate result was a trial to determine whether a single infusion of a 0.8 mg/ml alendronate sodium (pH 2.0) into a dog esophagus could cause irritations of the esophageal mucosa and, if so, whether the lesion completely resolved in seven days (to support one a week dosing in patients with 35 mg and 70 mg). The future plan was to treat dogs similarly once a week for four weeks to determine potential cumulative esophageal damage.
49 On 21 May 1997 the planned meeting took place between representatives of Lunar and Merck. It was attended by Yates, who went directly to it from the TPAC meeting. There is a sharp conflict as to what occurred. For reasons I will discuss later, I am satisfied that the Lunar News references to an alternative dosage regime for alendronate for the treatment of osteoporosis were present to the mind of Yates at the time of the meeting, and that the topic was expressly discussed at the meeting.
50 On 20 May Peter Browne sent an email to Yates as follows:
‘From: Browne, Peter
To: Yates, A. John
Cc: Senich, Barbara A. (WS)
Subject: Weekly-dosing
Date: Tuesday, May 20, 1997 5:12PM
John,
Congratulations again on the TPAC presentation, which I think was a great success for the Commercialization Team.
One item for follow up on the weekly dosing though. Can we send a quick note to the CT to remind people to keep the lid on the various weekly programs? I really want to keep this quiet, since it could be quite a bold stroke, and I don’t want anyone outside of Merck to know any earlier than they should, to preserve our lead in timing. I don’t think that TPAC people need to be reminded, but if you think that’s wise too, maybe we could communicate that to the TPAC too (maybe via Bob Bissett).
Regards,
– Peter’
The reply on the following day was:
‘From: Yates, A. John (5/21/97)
To: Browne, Peter
CC: Joanne Giesser, Senich, Barbara A. (WS), Daifotis, Anastasia G.
Mail*Link® SMTP RE>Weekly dosing
Peter
I agree and have instructed the folks in clinical research to keep mum (i.e. quiet) about this. Please send a note to the CT.
We should see animal data from CP Peter in about 2 weeks. In the mean time we (Anastasia, Jody Giesser and I) will work on putting a patent together.
John’
51 The dog studies were performed between 27 May 1997 and 6 June 1997 and were satisfactory so far as proof of the hypothesis was concerned.
52 On 6 June 1997 Edward Scolnick, President of Merck Research Laboratories sent an email to Yates and Elizabeth Stoner as follows:
‘From: Scolnick, Edward M.
To: Stoner, Elizabeth; Yates, A. John
Cc: Spector, Reynold
Subject: fosmax once or twice a week
Date: Friday, June 06, 1997 4:00PM
Priority: High
john liz, when w will we actaully start these studiesie FPI? I think in the competitive world we live in it isimportant we really try to do this project. More important than periodntal disease. / ed scolnick’
The reply of 9 June from Yates was as follows:
‘From: Yates, A. John (6/9/97)
To: Stoner, Elizabeth, Scolnick, Edward M.
CC: Anthony Sabatelli, Peter, Chennekatu P., Spector, Reynold, Daifotis, Anastasia G.
BCC:
Priority: Normal
Ed
I agree that this approach may well be very important to us.
Last week I had heard from CP Peter that the dogs exposed to ALN 0.8 mg/ml (simulating a high weekly dose) at pH 2 and sacrificed at 48 hours had only mild esophageal irritation. Another group of dogs are to be sacrificed at 7 days and the results from these are not yet available (they should be this week). If these look clean, then I think we have a very good rationale for thinking that a weekly dose may actually be safer than daily dosing with the same cumulative amount of ALN.
We are looking carefully (and quickly) at the patentability of weekly dosing. There are several intermittant [sic] regimens for bisphosphonates that have been included in patents, but Anastasia and I believe that we have at least some chance to get exclusive patent rights to use bisphosphonates weekly to lower the potential for esophageal ulceration (as well as enhance convenience).
Once we have the dog data and a better idea of the patentability (or whether any competitor’s patent might preclude us from using this approach) we will be in a position to make a Go/No Go decision on a clinical program. The clinical program itself would be quite simple. We will keep you updated. Thanks.
John’
53 The following exchanges of emails took place on 12 June 1997:
‘From: Browne, Peter
To: DiCesare, Elizabeth A.
Cc: Barbara Senich; Katdare, Ashok V.
Subject: Today’s Alt Form Task Force
Date: Thursday, June 12, 1997 10:32AM
Priority: High
Beth,
Some quick thoughts on today’s Alternative Formulations Task Force meeting:
Marketing is in agreement with HHPAC decisions on dosing/formulations. For each of the options, I’ll list our thoughts:
· Once weekly dosing – Very promising, important enough to start in clinical ASAP if we go with 40mg tablet, however, we recommend a 35 mg dose to make registration simpler (more directly substitutable for 5 mg and 10 mg given daily.
…
For the new product image, we will be doing the forecasts and marketing pros/cons in preparation for the results of the market research, to support the decision which will need to be made after full market research results are received. As you know, this is the one option that we can place into the market faster than risedronate and possibly tiludronate. Timelines discussed at recent DDST indicate that 1Q99 is fastest possible date. ANYTHING that can be done to accelerate this timing will be a major victory. I am assuming that we can gain internal sign-off by late August, or early September at the latest.
For once/twice weekly dosing, WHHM strongly supports the safety assessment program in dog model, and as stated earlier, we’d like to see 35mg over 40mg. We’d also like to file with 6 months BMD, if this has a REASONABLE probability of success. What are the tradeoffs in sample size, prob of success, etc. …’
(emphasis added)
‘From: Santora, Arthur C.
To: Holland, Sherry D.
Cc: DiCesare, Elizabeth A.
Subject: MORE bioavailability studies for FOSAMAX?
Date: Thursday, June 12, 1997 4:17PM
Sherry,
After you left the Alternative Formulations meeting today a topic was raised about the need to look at the bioavailability of a 70 or 80 mg dose if that were chosen for the new program. Beth and I recalled that the Bioavailability Dose-proportionality study went up to 80 mg. Ashok thought it was 40 mg. Either way, the biobatches only went to 40 mg, so a complete CMC package would be required. Your opinion is needed on whether a bioavailability study would be needed for a 70 or 80 mg dose if we chose to register the dose. The plan for clinical development includes a comparison to the 10 mg dose administered daily (our standard) so there would be data to support an equivalent BMD and bone biochemical effect of 10 daily and 70 or 80 weekly. Would any additional bioavailability studies be needed to support the application?
Art’
‘From: Holland, Sherry D.
To: Santora, Arthur C.
Cc: DiCesare, Elizabeth A.
Subject: RE: MORE bioavailability studies for FOSAMAX?
Date: Thursday, June 12, 1997 4:53PM
Art: What 70- or 80-mg dose? for what program? Last I heard we were looking at 35- or 40-mg dose 1x/week for prevention and 2x/week for treatment. The dose proportionality study with capsules went up to 80 mg. The FMI dose proportionality with the tablet went up to 40 mg (35 mg not included, but would arguably be in that range). The 35-mg tablet would probably require a PK study vs a registered dose (10 or 40) to confirm its performance (i.e. dose-adjusted GMR of AUC within (0.8, 1.25). To register a 70- or 80-mg tablet would require a PK study compared at least to 10 mg (also 5?, 40?) with the same type of relative bioeavailability [sic] question and perhaps also dose-proportionality. Are you looking at 70- or 80-mg once a week? Or 35- or 40-mg twice a week? Sherry’
‘From: Santora, Arthur C
To: Holland, Sherry D.
Cc: DiCesare, Elizabeth A.; Katdare, Ashok V.
Subject: RE: MORE bioavailability studies for FOSAMAX?
Date: Thursday, June 12, 1997 6:47 PM
Sherry,
Osteoporosis treatment. The potentially patentable claim is that the same total dose for alendronate (and related bisphosphonates) given once a week is less toxic to the esophagus (at least in dogs) than one-seventh the dose given once a day. Of course, it would be similarly efficacious. It would be less desirable to use 35 or 40 mg twice a week than to use double the dose once a week. Twice-a-week dosing would also greatly complicate the convenience claim (“I thought you said only one day a week,” remarked the lady with the kyphosis.) and muddle the patent claim.
Nobody could recall anyone actually “killing” a 70 or 80 mg dose at the T-pac or technical review. Although it may by [sic] “killed” at the next higher level review, its still in for now - - along with 35 to 40 twice a week in treatment.
Thanks for the info.
Art.’
(emphasis added)
54 The following exchange of emails took place on 13 June 1997.
‘From: Katdare, Ashok V.
To: Kloss, Michelle W.; Chen, Tzyy-Show; Porras, Arturo G.; Shuster, Judy; Doherty, Stephen J.; Gertz, Barry J.; Holland, Sherry D.; Jackson, Donna K.; Santora, Arthur C.; Bailey, Keith M.; Kramer, Ken
Cc: Philipson, James; Browne, Peter; Gardner, Colin R.; Senich, Barbara A. (WS); DiCesare, Elizabeth A.; Yates, A. John; Brooks, Marvin A.; Reynolds, Scott D.
Subject: Fosamax once-a-week or twice-a-week
Date: Friday, June 13, 1997 8:48AM
To all,
The alternative formulation task force had a good discussion on the subject topic yesterday. It was particularly useful since several different interpretations of the HHPAC discussion were presented. Marketing have expressed a strong interest in having a 35 or 70 mg tablet for once-a-week administration. It is believed that this approach may further realize a patent advantage.
I will present some additional ideas to cover these (ideas since yesterday’s meeting)
- A 35 mg tablet could be easily developed similar to current marketed formulations ie 35mg alendronate in 200 mg tablet weight. This tablet could be easily bracketed by stability database for 10 mg and 40 mg tablets. Although the 40 mg tablet is not filed in many ROW countries, the data could be used (I hope) to support quicker registration of the product. Obviously, a minimal data on the actual 35 mg could be generated with a commitment to follow long term data. This approach should not raise any bio issues.
- A 70 mg tablet has two following options -
a) A new tablet could be developed with 70 mg alendronate in 200 mg tablet weight. The drug to excipient ratio(s) for this tablet will be outside of our historical experience (Arturo – Please confirm this. This may have been looked at prior to my involvement in the project). If so, we will require some assurance of its biopharmaceutical performance. This could be done in parallel to Phase II studies with moderate risk. Obviously the recommendation will be [sic] assess this aspect prior to initiation of Phase III studies to eliminate the risk of tablet with non-optimum biopharmaceutical properties.
b) A second (and in my opinion preferred) option will be to develop a weight-multiple of the 40 mg tablet ie 70 mg in 350 mg tablet weight. Although the tablet will be somewhat larger in this case, it could be supported by existing stability and bioavailability data. Phase III studies could be initiated without having to assess the biopharmaceutical performance of this tablet. Although additional stability data will require to be generated, hopefully filing could be achieved with minimal data with supportive data form [sic]40 mg tablets (Judy, Ken, Michelle, Steve, Donna – please confirm).
Obviously one caveat is that Marketing and MRL management will have to agree to slightly larger tablet for 70 mg and most importantly use of 70 mg once-a-week. The ongoing safety studies ill [sic] help us make that decision.
Please call if you have any questions, ashok’
(emphasis added)
‘From: Yates, A. John
To: Kloss, Michelle W.; Chen, Tzyy-Show; Peter, Chennekatu P.; Porras, Arturo G.; Stoner, Elizabeth; Shuster, Judy; Doherty, Stephen J.; Gertz, Barry J.; Holland, Sherry D.; Jackson, Donna K.; Spector, Reynold; Daifotis, Anastasia G.; Santora, Arthur C.; Bailey, Keith M.; Katdare, Ashok V.; Kramer, Ken; Sabatelli, Anthony
Cc: Philipson, James; Browne, Peter; Gardner, Colin R.; Senich, Barbara A. (WS); DiCesare, Eliazbeth A.; Brooks, Marvin A.; Reynolds, Scott D.
Subject: RE: Fosamax once-a-week or twice-a-week
Date: Friday, June 13, 1997 9:25AM
Ashok
I agree with the approach you outlined here. A 70 mg tablet in a 350 mg weight tablet would probably be acceptable as the size is still quite small, especially if it is made to be close to spherical, so there would be no question concerning increased likelihood of delayed esophageal transit.
My feeling about once vs. twice per week is that once per week is preferable if we can support the rationale that esophageal irritation is more likely with more frequent dosing and that a one week off treatment phase (i.e. weekly dosing) allows recovery of any irritation that is seen. It would also be more convenient for patients.
Two counterarguments to this are:
1) There may be greater variability in absortion [sic]. Thus the % absorbed with a single daily dose varied widely in the PK studies, but was not consistently low or high within individual patients. Therefore, the average bioavailability over a 1-2 week period is likely to be less than a single day’s variation. However, we know that even three monthly dosing with IV bisphosphonates can be effective, and thus I do not think that this effect will have much practical importance in terms of variability of response.
2) The dose in [sic] in excess of the 40 mg dose which is the maximum that we have significant experience with (although 80 mg/day daily for 3–6 months was well tolerated in Paget’s patients). Although this is a natural reaction, I think we can trust the dog esophagus model to be informative about what will happen in humans, so that if weekly dosing is less irritating than daily dosing with the same cumulative dose, we should be able to expect greater safety in humans with the weekly 70 mg dose.
I was informed by CP Peter that the 7 day results from the high single dose study should be available early next week.
John’
(emphasis added)
‘From: Santora, Arthur C.
To: Kloss, Michelle W.; Chen, Tzyy-Show; Peter, Chennekatu P.; Porras, Arturo G.; Stoner, Elizabeth; Shuster, Judy; Doherty, Stephen J.; Gertz, Barry J.; Holland, Sherry D.; Jackson, Donna K.; Spector, Reynold; Daifotis, Anastasia G.; Yates, A. John; Bailey, Keith M.; Katdare, Ashok V.; Kramer, Ken; Sabatelli, Anthony
Cc: Philipson, James; Browne, Peter; Gardner, Colin R.; Senich, Barbara A. (WS); DiCesare, Elizabeth A.; Brooks, Marvin A.; Reynolds, Scott D.
Subject: RE: Fosamax once-a-week or twice-a-week
Date: Friday, June 13, 1997 11:33AM
FYI
An 80 mg dose in a 200 mg image tablet was studied as the top dose in a Phase I bioavailability dose proportionality study (in about 1990–1) and the fractional absorption was constant across the dose range studied. A 80 mg dose was not included in the definitive biobatch bioavailability study.
Art’
(emphasis added)
55 An Alendronate Sodium Esophageal Irritation Study in Dogs was designed to support a once a week tablet in patients (35 mg and 70 mg tablets) by infusion at a rate equivalent to 80 mg. The trial commenced in July 1997.
56 At some stage prior to 17 July 1997 Yates instructed Daifotis to complete a Confidential Memorandum of Invention (CMI) in relation to the proposed dosage regime. I discussed that topic in Arrow Pharmaceuticals Limited v Merck & Co., Inc. [2004] FCA 1131. Yates initially claimed that this was done in May, but I do not accept that evidence. It is most likely that it was done sometime after 6 June. The CMI had to be approved and countersigned higher up the chain of command before submission to the Patent Department, which took place on 17 July 1997.
57 Between May and July 1997 market research was conducted by Merck in respect of dosing options which was reported upon on or about 20 August 1997. The report of that research makes clear that the driving force behind the marketing push was the need to counter the competitive threat posed by the entry of risedronate onto the market. The study tested (among other things) the acceptance of 40 mg twice a week.
58 On 22 July 1997 US provisional application 60/053,351 was filed but later abandoned. Provisional specification 60/053,535 was filed on 23 September 1997.
59 In September 1997 Yates told a closed symposium that Merck were to conduct once weekly dosage clinical studies with alendronate. The trial was conducted with favourable results and was reported upon in an article by Schnitzer et al (including Yates) published in 2000.
60 On 29 December 1997 Elizabeth DiCesare of Merck encouraged members of the Fosamax Commercialisation Team Meeting to seek out documentation on once weekly dosing that pre-dated the publication of the April 1997 edition of Lunar News, obviously for patent purposes. The internal emails in early August 1996 came to light as a result of that request.
61 On 5 March 1998 a meeting request and background package was forwarded by Merck to the FDA concerning the proposed clinical trial. That package included the following (omitting footnotes):
‘Clinical Studies: Upper GI Safety of Alendronate 40 and 80 mg Daily
While there are no new studies of 35- and 70-mg doses of alendronate, similar high doses of alendronate were studied in the Paget’s disease development program. A 40-mg daily oral dose of alendronate was used in the pivotal Phase III Paget’s disease studies and oral alendronate treatment at dosages up to 80 mg daily for up to 6 months was included in two Phase II studies. Although the exposure of Paget’s disease patients to alendronate 80 mg daily was limited (36 patients for 6 months), this dose was generally well tolerated and no previously unrecognized adverse effects were found. In the total Paget’s disease treatment experience, no patient discontinued treatment with alendronate due to a serious drug-related adverse experience. Overall, in all Paget’s disease studies the discontinuation rate due to a clinical adverse experience was 5.6% for alendronate-treated patients. Additional experience on the use of daily oral alendronate 40 mg is provided from a Phase II osteoporosis treatment dose-ranging study in which patients received oral alendronate 40 mg daily for up to one year. Upper gastrointestinal adverse experiences (none of which was serious) resulted in discontinuation in about 10% of these osteoporotic patients (no placebo or alendronate 10 mg patients withdrew from therapy due to drug-related upper gastrointestinal adverse experiences).’
(emphasis added)
62 On 16 April 1998 Yates made a presentation to the FDA. The overheads included the following:
‘Evidence for Efficacy
1. Rats given same total dose of ALN in 2, 4, or 8 s.c. inj./month after OVX for 12 weeks achieved comparable increases in BMD
2. OVX baboons treated with ALN 0.05 or 0.25 mgP/kg IV q 2 wks for 2 yrs had reduced bone turnover and increased bone [mass and bone] strength
3. Postmenopausal women treated with ALN 10 mg IV q 3 months increased spine and forearm BMD over 1 year vs. controls
Therefore, data from rats, baboons and humans suggest similar efficacy with less frequent vs. daily dosing.
1. Seedor, JBMR, 1991; 2. Balena, JCI, 1993; 3. Passeri et al. Bone and Mineral, 1991’
(handwritten annotations in brackets)
‘Evidence for Safety
Clinical Trials
· Paget’s disease treated with 80 mg/day for 3 to 6 months in 42 patients with good tolerability (no discontinuations due to UGI AE)
· 40 mg dose is generally well tolerated in Paget’s disease, although some excess of discontinuations due to nonserious UGI AEs in postmenopausal osteoporosis population
Marketed Use
Few reports of GI AEs from marketed use of 40 mg’
63 On 17 July 1998 an application for Australian Patent AU 84936/98 was lodged but lapsed. The application for European Patent EP 0 998 229 B1 was made on the same day.
64 In early August 1998 the Australian Drug Evaluation Committee resolved to advise that the induction of Alendronate Sodium (10 mg and 40 mg Fosamax tablets) should be extended to the treatment of osteoporosis and Paget’s Disease of the Bone.
65 On 28 August 2000 Australian Patent AU 741818 was filed. The priority date claimed for Australian Patent No 741818 was 2 September 1999.
66 On 17 November 2000, 70 mg once weekly Fosamax received TGA approval in Australia for export.
67 On 7–8 December 2000 the Australian Drug Evaluation Committee resolved to approve (i) the 70 mg once weekly regimen for Fosamax for the treatment of post-menopausal osteoporosis and (ii) the amendment of product information for 5 mg, 10 mg and 40 mg tablets.
68 On 9 February 2001 70 mg once weekly Fosamax received TGA approval for use in Australia.
69 In May 2001 70 mg Fosamax tablets received PBS listing in Australia.
FURTHER FINDINGS
Lunar Meeting
70 The Lunar meeting has relevance because of the question as to what Yates knew of the suggestions in Lunar News about less frequent and higher dosing of alendronate. He denied being aware of those suggestions until July 2001. I have already referred to internal documents showing certain of the articles having been circulated to him. All of the Lunar participants in the meeting gave evidence and were cross-examined. Yates was the only one of the four Merck attendees to give evidence. I accept the substance of the evidence of the Lunar participants in preference to that of Yates. None was significantly shaken in cross-examination. Attention was drawn to some discrepancies between the evidence given and prior depositions but I was satisfied by the explanations given.
71 On the other hand, the version given by Yates stretches credulity. He accepts that he attended the meeting by prior arrangement as the technical expert on alendronate. He acknowledges that there was an agenda item ‘misrepresentation of alendronate’. He says he had no background documentation. He says he did not read the Lunar News articles referring to alendronate before the meeting and was not briefed with any other documents or correspondence. He says that he was unaware of any issue concerning dosage. He claims that he thought the issue related to representations as to lack of safety.
72 I am satisfied that:
(1) In September 1996 Sherwood of Merck had objected to Mazess about 40 mg ‘off label’ dosing being suggested in Lunar News.
(2) Copies of relevant issues of Lunar News were in the room at the time of the meeting.
(3) The reference to once weekly dosing at a higher rate as per the July 1996 Lunar News article was discussed at the meeting and Sherwood was sharply critical of Lunar Corporation in that respect.
(4) Lunar agreed to provide Yates with advance copies of Lunar News to comment on any articles on bisphosphonates.
73 I am satisfied that Yates’ evidence should be rejected and find that he was well aware of the contents of the articles published in Lunar News up to the time of the meeting that had mentioned alendronate.
Conception of alleged Invention
74 I do not accept that Yates conceived of the alleged invention in April 1996 after receiving the results of the dog trials. Yates has little credit in view of his evidence concerning the Lunar News meeting. He did nothing concrete about any observations he made immediately following April 1996. He is not corroborated by any other alleged inventor. The reaction of Yates and Santora when the issue was raised in August 1996 is plainly inconsistent with the claim that is now made. Yates noted the result of the dog trial, but not in a manner consistent with the alleged invention having been made at the time claimed. It will be recalled that his reaction was:
‘Possible increased GI toxicity because of high dose. However, this is speculative because single dosing in dogs was not a problem, so perhaps the mucosa can rebound if given 6 days without alendronate. This would need to be tested in the dog model.’
75 Further, his reaction when the matter became live in 1997 is inconsistent with any invention having been made in April 1996. I am satisfied that his evidence concerning April 1996 was an ex post facto rationalisation, which further diminishes his credit. I am reluctant to accept his evidence where it is not corroborated by documents or other reliable evidence.
76 The first clear mention of once a week dosing at 40 mg or 80 mg is in the July 1996 edition of Lunar News. That was followed in early August 1996 by the suggestion in the internal Merck documents that Italian investigators were looking into dosing at 40 mg once per week to ‘get around the daily restrictions and potential for AEs with the 10 mg dose’. That led to no action by Merck and there is no evidence of any recommendation for action at that time from Yates or Santora. The next relevant reference was in the April 1997 edition of Lunar News to (inter alia) a dose of 40 mg once per week.
77 The first clear reference in the internal Merck documents after August 1996 was on 7 May 1997 in the email from Santora to Peter. It will be recalled that that referred to a hypothesis that:
‘a relatively high dose might be well tolerated, even in a patient with esophageal reflux, if it were administered weekly, with time between doses for recovery from any damage which might occur with any one dose.’
Things moved quickly thereafter. It is apparent from the internal Merck documents that the marketing side was pushing strongly for both alternative formulations and alternative dosing regimes. A decision to proceed was made at the meeting on 20 May 1997. Yates prepared the relevant part of the report that was considered at that meeting and he and Santora spoke to the proposal. I have set out relevant portions of the report above and do not repeat them here.
78 Based upon the evidence as a whole, particularly the contemporaneous documents of Merck, some conclusions can be drawn. There was low level consideration of the possibility of less frequent administration of alendronate at a higher dose for the prevention and treatment of osteoporosis from August 1996 onwards. The same point had emerged from the Lunar News articles that reached the attention of relevant Merck personnel. The low level of interest was probably because of lack of impetus from the marketing department. There was a pricing problem if one pill was substituted for seven. By May 1997 considerable marketing interest had developed in the possibility of an alternative dosing regime leading to some more serious attention being devoted to it by the research department. There was never any real doubt that a higher and less frequent dose of alendronate would be efficacious in the prevention and treatment of osteoporosis due to the known long half life of the drug. There was also never any real concern that such a regime would lead to any significant increase in GI side effects, principally (but not only) because of the experience gleaned through the administration of higher doses for the treatment of Paget’s disease. What Yates added when he focused upon the issue was his recollection of the results of the 1996 dog trials which he recognised might provide a scientific explanation for the relative GI safety of the higher and less frequent dosage and which could possibly form the basis for a patent. Whether or not that explanation was correct and whether or not it could lead to a patent, clinical trials would still be required and could be justified (as they were) on the basis of the experience to that time including the Paget’s experience. The possibility of patent protection extending beyond the life of the existing patents was a most desirable side effect of the exercise and encouraged Merck to devote resources to the project, although the existence of a new patent was not in itself necessary for FDA approval to be obtained. It is also clear from the Merck documents that the focus of the Merck personnel was upon 40 mg and 80 mg doses and that 35 mg and 70 mg doses were not distinguished from them in any practical technical sense. To the extent that any evidence of Yates was to the contrary of these conclusions I reject that evidence.
79 I am not, however, satisfied that the genesis of the alleged invention lay in the articles in Lunar News. There is no mention of them in the relevant internal research reports. The interest of Italian researchers was known at about the same time. The possibility of alternative dosage regimes was ‘on the table’ at Merck during 1996. As it happened the issue was of no great interest until the marketing impetus developed in 1997. The idea is not so novel as only to be explicable as having been taken from the suggestion in Lunar News. Given the known compliance problems, it was an alternative clearly worth considering.
GROUNDS OF INVALIDITY
INVENTION – MANNER OF MANUFACTURE
80 Of the seven claims which are now in issue, three are expressed to be method claims (claims 1–3) and four are expressed to be composition claims (claims 5, 6, 9 and 10). It is tolerably plain on the face of the patent that the so-called composition claims lack subject matter. There is nothing that is claimed to be new or novel with any utility about the carrier, the active ingredient, or the quantity or the method by which the ingredients are ‘composed’. Reference is only made to standard mixing and formulation techniques. In particular, there is no practical meaning or substance to the statement that the composition is ‘adapted for oral administration as a unit dosage according to a continuous schedule having a periodicity of about once weekly’. It is not suggested that there is any problem with an oral composition of the type and size identified being used as a once weekly dose. There is nothing about the composition itself that ‘adapts’ it to once weekly dosing as compared with any other dosing. These are not composition claims as that concept would normally be understood, that is, claims to a new and unique compound. They are not combination claims whereby the whole is something different from the sum of the parts. When properly analysed the composition claims are devoid of practical content. They are not ‘inventions’ and are not a manner of manufacture.
81 The relevant claims of this patent will stand or fall by the so-called method claims. The disclosures in the specification include the following. It was known that alendronate monosodium trihydrate on an alendronic acid active basis was effective in preventing and treating osteoporosis in a human. It was known that it could be orally administered, that is, taken by mouth in various amounts and at various time intervals. It was known that oral administration of the compound had the potential to give rise to GI side effects in a small but significant proportion of the population. It was known that there could be problems of compliance with a daily dosage regime of the type described in the specification. Thus, in substance, each claim relates to the use of a known substance with known properties for a known purpose in a known manner. There is no claim of any new process or method of administration as such. There is no claim of discovery of new physical properties in the compound.
82 It is trite law that a mere new use for an old thing is not patentable. However, a discovery that an old substance may be so used as to produce a new result may possibly give rise to a patentable invention. The old substance in that case is treated as if it were new, the previously unknown or unsuspected qualities of it being revealed by the discovery if the discovery in question is a consequence of scientific ingenuity. The principle extends to a process that results in a new and useful effect that is an artificially created state of affairs of economic utility (National Research Development Corporation v Commissioner of Patents (1959) 102 CLR 252 (NRDC) at 265, 277; Wellcome Foundation Ltd v Commissioner of Patents (1980) 145 CLR 520 at 528).
83 However, a process claim cannot lead to a monopoly in a substance limited to its use in the process. The process makes no contribution to the substance – it takes advantage of its properties. The substance is merely an ingredient in that process (Wellcome Foundation Ltd v Commissioner of Patents at 529–530). As Parker J said in Adhesive Dry Mounting Co Ltd v Trapp & Co (1910) 27 RPC 341 (at 353):
‘The idea of using an old material for an entirely new purpose, not being analogous to purposes for which it has theretofore been used, may be good subject matter, but such idea, however ingenious, can hardly justify a claim for the material itself.’
84 As was said in Commissioner of Patents v Microcell Ltd (1959) 102 CLR 232 (at 249):
‘Many valid patents are for new uses of old things. But it is not an inventive idea for which a monopoly can be claimed to take a substance which is known and used for the making of various articles, and make out of it an article for which its known properties make it suitable, although it has not in fact been used to make that article before.’
85 The authorities which were analysed in Commissioner of Patents v Microcell Ltd at 247–249 including Re B.A.’s Application (1915) 32 RPC 348 in which it was said (at 349):
‘once a substance is known, its methods of production ascertained, its characteristics and its constituents well defined, you cannot patent the use of that for a purpose which was hitherto unknown.’
86 That line of authority was recently applied in the High Court in NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd (1995) 183 CLR 655 which in turn approved the Full Court decision in NV Philips Gloeilampenfabrieken v Mirabella International Pty Ltd in this Court (1993) 44 FCR 239. No doubt is cast upon that line of authority in Advanced Building Systems Pty Ltd v Ramset Fasteners (Aust) Pty Ltd (1998) 194 CLR 171 (see eg 191–192).
87 Guided by those authorities, and having regard only to the disclosures in the specification itself, I would hold that each of the so-called method claims was one way of utilising alendronate and its known qualities for the known purpose of preventing or treating osteoporosis by a known method of oral administration. They are in the nature of directions for use. That does not constitute an invention or a manner of manufacture.
88 I am reinforced in that conclusion by considering the practical implications of the contrary view in the field of pharmaceutical chemical compound patents, well-illustrated by the present case. Merck acquired the base patents involving the alendronic acid and pharmaceutical compositions containing it which disclosed administering the same for the purpose of inhibiting bone resorption. That patent would have prevented or hindered any clinical trials being conducted otherwise than with the licence of the patentee in those countries in which a patent had been granted. Leaving aside the legal effect of the patent, only the patentee (or licensee) would have the commercial motivation to proceed with the substantial trials required. The regulatory regime is such that clinical trials are essential before there can be commercial exploitation. The opportunity for refining and improving the application of the base patent is, in a practical sense, limited to the patentee or those authorised by it. Once Merck obtained the base patent, it could control that field. As it controls use of the compound, it acquires the most widespread knowledge of the application of the compound. The patentee will thus have a virtual monopoly of the commercial development of it. In the present case, that led to the trihydrate salt patent, which had a life extending well beyond the life of the base patent in other jurisdictions. Certain of the claims of that patent would arguably include the method claims in issue here. The patentee has a practical monopoly of the opportunity of further refining the use of that invention. It would be a surprising result if using that monopoly and the information received from clinical trials of the compound would enable a further refinement of the necessary instructions for use of the compound to be itself patentable subject matter. This would extend the practical life of the patent by conferring a monopoly over the best method found in practice to put it into effect. There is something anomalous about a patent being obtained for all pharmaceutical uses of a chemical compound without disclosing any particular dosage regime for any particular use but with the patentee later claiming a new, stand-alone, patent for a particular dosage regime for a particular purpose that was contemplated at the time of the base patent, with no new properties of the compound having been discovered in an inventive fashion in the meantime.
89 Every pharmaceutical drug requires a dosage regime (or regimes) for practical prescription. Here, Merck, having the trihydrate salt patent, proposed particular dosage regimes for the prevention and treatment of osteoporosis and Paget’s disease to the regulatory authorities. Approval was given. That led to the required process of clinical trials which in turn led to marketing approval for the particular regimes. There is no evidence as to precisely how or why those original dosage regimes were chosen. Based upon the evidence concerning the development of the patent in suit, the choice is likely to have been very strongly influenced by marketing considerations. No patent was claimed for those dosage regimes. If those regimes had been known at the time of the salt patent, they should have been disclosed as part of the consideration for the grant of the patent (s 40 of the Act). The GI side effects that were disclosed once Fosamax was used by the public were regarded by Merck as being of the order of magnitude that had been anticipated by reason of pre-existing knowledge and the experience in the clinical trials. Problems with compliance may have subsequently become more obvious. The claims in suit simply alter the dosage regime that Merck itself had proposed and introduced. Furthermore, the decision to do this was driven by a marketing desire to head off inroads into the market by risedronate. The proposal did not emanate from the scientific or research area of Merck, which only responded to the suggestion. In my opinion, such an alteration in Merck’s own regime does not amount to patentable subject matter as such. Even if it could qualify as a patent of addition pursuant to ch 7 of the Act it would have been limited to the term of the salt patent.
90 Indeed, it is surprising that in this context there has been no discussion of ch 7 of the Act dealing with patents of addition. The legislature obviously intended that, even if what is proposed is a true improvement in or modification of the main invention, and not merely instructions as to use of it, a consequent patent of addition should not extend beyond the life of the base patent. (See generally Terrell on the Law of Patents, 12th edn, pars 133–141.)
91 However, it is submitted on behalf of Merck that the conclusion which I have reached is precluded by the decision of the Full Court in Bristol-Myers Squibb Co v FH Faulding & Co Ltd (2000) 97 FCR 524 (Faulding). The background to that decision is succinctly summarised in the judgment of Black CJ and Lehane J (at [1]–[4]):
‘This appeal concerns two petty patents for methods of administering taxol. Taxol has been known, for about three decades, to have anti‑carcinogenic properties. It inhibits the division of cancer cells. Other drugs used in the treatment of cancer have that effect also; taxol, however, does so by a mechanism that differs from the way in which other drugs inhibit cell division. Consequently, taxol has for many years been recognised as potentially efficacious where other drug treatments have failed.
There are, however, considerable difficulties with the use of taxol in the treatment of cancer. One difficulty is its scarcity. It is a naturally occurring compound extracted from the bark or the needles of the western, or Pacific, yew. Extraction is a slow process and taxol is not plentiful. Secondly, taxol is relatively insoluble in water and is administered (there was no evidence of any other possible mode of administration) in a mixture of Cremophor EL and dehydrated ethanol. Thirdly, taxol is highly toxic: among its side effects are toxicity of the blood (particularly, neutropenia) and of the nervous system (peripheral neuropathy). Additionally, the administration of taxol was found frequently to produce hypersensitivity reactions, often severe reactions: this may have been due either to the Cremophor or to the taxol itself.
…
The claims of the earlier patent were:
“1. A method for administration of taxol to a patient suffering from cancer comprising infusing from 135 to 175 mg/m2 of taxol over a duration not exceeding 6 hours.
2. The method of claim 1, wherein said administration comprises infusion of 135 mg/m2 of taxol.
3. The method of claim 2, wherein the duration of said infusion is not greater than 3 hours.”
The claims of the later patent were:
“1. A method for treating cancer in a patient suffering therefrom including infusing from 135 to 175 mg/m2 of taxol over a duration less than 6 hours wherein said method results in a reduction of hematological toxicity and neurotoxicity compared with infusing greater than 170 mg/m2 of taxol over a duration of 24 hours.
2. A method according to claim 1 wherein said method includes infusing 175 mg/m2 of taxol.
3. A method according to claim 1 or claim 2 wherein said method includes infusing said taxol over a duration not exceeding 3 hours.”’
92 All members of the Court affirmed the proposition that a method of medical treatment of the human body is patentable, following in that respect the decision of a previous Full Court in Anaesthetic Supplies Pty Ltd v Rescare Ltd (1994) 50 FCR 1. Arrow challenges that finding but accepts that I am bound to apply it.
93 The next topic considered by Black CJ and Lehane J was whether what was claimed was a manner of new manufacture. It had been held by the primary Judge that it was clear from the specification that the requirement of invention or inventiveness was absent. It was not merely a claim for a new use of an old substance but a claim for the same use of an old substance. Furthermore, it was held that the specification disclosed that the claimed inventions were the product of routine testing which verified a hypothesis arising from analysis of reports of earlier trials. Black CJ and Lehane J disagreed with that reasoning. The essence of their disagreement is to be seen in the following passage (at [31]):
‘The findings of the trial judge as to the failure of the claims to meet the threshold requirement of inventiveness relied, as to what was known and as to the studies leading to the claimed invention, only upon what is disclosed in the specifications. The specifications reveal, as his Honour pointed out, that both the efficacy of taxol as an anti‑carcinogenic (particularly in relation to drug‑refractory ovarian cancer) and the mechanism of its action were known. His Honour found, accordingly, that the claimed invention was not merely a claim for a new use for an old substance (his Honour’s shorthand) but a claim for the same use of an old substance, thus failing the Microcell test. In our opinion, however, that formulation overlooks two things. One is that the claim is for a method, not a product; the other is the importance of the phrase “nothing but” in the Microcell principle: as to both points, see NRDC, especially at 262. Taxol may, if used in accordance with the claimed invention, be used for a purpose for which its known properties make it suitable; it does not follow that the method claimed does not involve an inventive step. Nor, if the method was proved to be efficacious by a routine process of trial and error (the authorities cited by his Honour have to do with how much, in order to destroy novelty, an anticipation must reveal), does it follow that the claimed invention is obvious or does not involve an inventive step: what matters is whether, to the skilled but unimaginative worker in the field, the claimed method was obvious in the sense that the worker, not necessarily seeing that the method was likely to be safe and efficacious, would have seen that it was one which justified investigation.’
94 In assessing the question for themselves, their Honours said (at [43]–[44]):
‘In order to determine whether the claimed invention had the necessary quality of inventiveness what must be asked, in our view, is whether in the light of the prior body of knowledge discussed in the specification, and given the desirability of treating patients as outpatients rather than admitting them overnight and, generally, of reducing cost and inconvenience, the skilled but non‑inventive worker in the field would have seen that infusion over a period less than six hours (particularly, over three hours) with premedication, of approximately the doses actually selected for the trial, was worth trying. In answering that question, it must be borne in mind that “trying” was plainly a process which would involve considerable effort on the part of a large number of people, much expense and the subjection of patients, already very ill, to a form of treatment which, while it might in some cases produce some remission, was known to have the potential to cause very unpleasant, and sometimes life‑threatening, side effects: the circumstances by no means resemble the example of the stainless steel sink referred to in Microcell at 248.
What can be gathered from the specification is that the previous teaching encouraged longer infusions (usually twenty-four hours, but certainly not less than six), doses at the upper end of, or above, the range claimed in the petty patents and premedication. Infusions of shorter duration had been tried, without premedication, and had been found unsatisfactory. Given that, frequently, hypersensitivity reactions occurred shortly after the commencement of an infusion, there was some doubt as to whether the infusion period was actually of much significance in relation to reactions of that kind. Nevertheless, it could not be said that there was anything in the teaching referred to which particularly encouraged a view that a three hour infusion of dosages within the range claimed would be safe and would work. The position was simply that the administration of taxol in that way had not (so far as the material referred to goes) been tried, with premedication, and there were several practical reasons why it was desirable to reduce infusion times if possible. Is the Court, armed with that knowledge, equipped with the hindsight against which authority warns (see Minnesota at 293) but otherwise on the basis of its own untutored understanding (particularly, without the benefit of expert evidence), to say that the claimed invention lacks inventiveness (would have been obvious to the skilled worker in the field)? In our view, we should be very slow to do so. If one puts aside the benefit of hindsight, how is the Court to know whether experts would have found what was tried obvious or, for reasons of which we know nothing, counter‑intuitive? It is important to remember warnings (see, for instance, CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 285) that “[the] Court should be careful to avoid assuming a technical expertise it does not have”. We are not prepared to hold, on the material before us, that the quality of inventiveness was lacking.’
Finkelstein J agreed with Black CJ and Lehane J in this respect.
95 It does seem to follow that it was held that a mere dosage regime of a known chemical compound for a known therapeutic use based upon known properties and involving no new method of administration was patentable. As will be apparent from the foregoing, I regard that conclusion as being difficult to reconcile with prior and binding authority if applied to the present circumstances. My reservations are underlined by considering the decision of the United Kingdom Court of Appeal in relation to taxol – Bristol-Myers Squibb Co v Baker Norton Pharmaceuticals Inc [2001] RPC 1 (Bristol-Myers). As pointed out by the High Court in Aktiebolaget Hassle v Alphapharm Pty Ltd (2002) 212 CLR 411, great care needs to be taken in reading United Kingdom cases in this field because of the significant differences which have developed. However, Aldous LJ analysed the claim (at [34]) as being:
‘for the use of two known products to produce a medicament with the novelty relied on being provided by the alleged new application.’
It was clear that, even under the extended European Community rules, novelty could only lie in a new therapeutic use. Aldous LJ said (at [42]):
‘… I turn to claim 1. The judge was right to conclude there was not a claim for a second therapeutic use. The medicaments in question were known to be suitable for treating cancer. The remainder of the claim relates to the way such a medicament was to be used. A similar conclusion was reached by the Dutch Court of Appeal in Bristol-Myers Squibb v Yew Trew of 25 June 1998.’
96 I have been referred to the English decisions corresponding to this case (Merck & Co., Inc’s Patents [2003] EWHC 5 (Pat); [2003] FSR 29 at first instance; and Merck & Co., Inc’s Patents [2003] EWCA Civ 1545; [2004] FSR 16 in the Court of Appeal) and to the corresponding United States decision (Merck & Co., Inc. v Teva Pharmaceuticals USA, Inc. 288 F. Supp. 2d 601). It is accepted by counsel that the law in each place is sufficiently different from our present law to distinguish those decisions and limit their utility for present purposes. However, a revealing exercise in the decision at first instance in the United Kingdom was counsel’s redrafting of some statements from Holman J at para [111] in the English decision in Bristol-Myers substituting alendronate for taxol, osteoporosis for ovarian cancer, adverse GI effects for neutropenia and ‘the new and old alendronate dosing regimes’ for ‘the new and old taxol dosing regimes’. The result is as follows (at 519):
‘In the present case, however, the drug alendronate is exactly the same; the method of administration, orally, is exactly the same; and the therapeutic application or purpose, namely the attempt to treat osteoporosis, is exactly the same. The only difference is the discovery that if the drug is administered in a unit dosage form of 70mg once weekly rather than 10mg once daily an undesirable side-effect, adverse GI effects, is less than it otherwise would be, whilst the therapeutic effect remains. No previously unrecognized advantageous properties in the chemical compound have been discovered … All that has been discovered … is that if the compound is administered once a week rather than daily, one of its disadvantageous side effects will be less than it otherwise would be.’
As Jacob J said, counsel for Merck found no answer to that. That analysis is consistent with that of Heerey J at first instance in Faulding (1998) 41 IPR 467 and with my analysis in this case.
97 I am, of course, bound by the ratio decidendi of the Full Court decision in Faulding. I am inclined to the view that Finkelstein J was correct in his opinion that a ground of decision by a Full Court binds a single judge, even though there were other grounds for the decision (Faulding [146]–[161]). In my opinion, however, Faulding can be distinguished on the facts in relation to the present issue. Taxol was a naturally occurring substance with known beneficial properties which could not be utilised without the invention. The dosage regime was the key that unlocked the door to utility. That was at least arguably akin to a first therapeutic use case along the lines developed in the United Kingdom (cf NRDC). That is not the case here. Further, it is arguable that the dosage regime in Faulding was the result of a newly discovered technical effect, namely the reduction in neutropenia. There is no responding newly discovered technical effect in the present case. Thus, in the absence of binding authority to the contrary, I find that each of claims 1, 2, 3, 5, 6, 9 and 10 is invalid. Although that is sufficient to dispose of the case, I propose to consider certain of the other grounds for invalidity that have been advanced on the part of Arrow.
Novelty
98 The combined effect of s 18(1)(b) and s 7(1) of the Act is to require a comparison between the invention so far as claimed on the one hand with the prior art base on the other to see whether the invention as claimed is novel – something new or different. The principal prior art information relied upon by Arrow consists of the Lunar News articles of April 1996, July 1996 and April 1997. For reasons not explored in argument, Australian patent 625704 was not relied upon by Arrow as an anticipation of the claims in suit. There is a threshold question as to whether each of those issues was a document containing information that was publicly available, whether in or out of Australia. I can infer from the evidence that copies of each of the relevant issues of Lunar News were distributed to persons in both the United States and Australia prior to the priority date, those persons being free in law and equity to make use of the documents and the information in them without restriction. It is submitted on behalf of Arrow that that satisfies the statutory test. On the other hand, leaving aside the Lunar website, there is no evidence that any of the relevant issues was available for inspection by members of the public at any library or could be purchased by members of the public or even supplied on request to members of the public. Further, such distribution as took place was to persons chosen by Lunar Corporation rather than simply to members of the public at random. It is submitted on behalf of Merck that such publication does not meet the statutory test.
99 There is a line of authority which establishes that, in this context, communication to a member of the public without any restraint as to the use of the information by that person amounts to making the information publicly available (Fomento Industrial SA v Mentmore Manufacturing Co Ltd [1956] RPC 87 at 99–100; Sunbeam Corporation v Morphy-Richards (Australia) Pty Ltd (1961) 180 CLR 98 at 111; Re Bristol-Myers Co’s Application [1969] RPC 146 at 154–155; PLG Research Ltd v Ardon International Ltd [1993] FSR 197 at 226; and (in a different context) Telstra Corporation Limited v Australasian Performing Right Association Limited (1997) 191 CLR 140 at 156–157, 158, 174, 199–200, 202). I agree with the submission on behalf of Arrow that this established line of authority is applicable to the present legislation.
100 The question remains as to whether distribution to persons selected for commercial reasons by the publisher is communication to a member of the public for the purpose of the application of this test. In my opinion, it is. The recipients are just as much members of the public as are persons who purchase subscriptions to magazines or buy newspapers or who are on a mailing list. I would exclude from that category Mr Davies, the agent in Australia of Lunar. However, Davies was provided with copies of Lunar News in order to distribute them and I accept that he did so.
101 On any view, the Lunar Corporation website would make information available to the public. I accept that the April 1996 issue of Lunar News was available on that website. There is no direct evidence as to the July 1996 and April 1997 issues. On the balance of probabilities I would infer that the system would continue and that each was available.
102 The question as to what is novel compared with the prior art base is governed by principles that have been long established in the High Court eg Meyers Taylor Pty Ltd v Vicarr Industries Ltd (1977) 137 CLR 228 at 235; Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 and applied in a number of Full Court decisions eg RD Werner & Co Inc v Bailey Aluminium Products Pty Ltd (1989) 25 FCR 565 per Lockhart J at 568–569; Nicaro Holdings Pty Ltd v Martin Engineering Co (1990) 91 ALR 513 per Gummow J at 528; CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 277–280; and ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc (2000) 106 FCR 214 at [40]–[51]. The reverse infringement test will generally be appropriate.
103 In my opinion there is no relevant link between the various Lunar News articles and each must be viewed individually.
April 1996
104 It will be recalled that the article included the following:
‘An intermittent treatment program (for example, once per week, or one week every three months), with higher oral dosing, needs to be tested.’
105 In my opinion, that is a clear anticipation of claim 3, subject to the effect of the words ‘needs to be tested’. The gist of the novelty in claim 3 is a continuous schedule of oral administration having a dosage interval which is once weekly. The attempt on behalf of Merck to read 35 mg or 70 mg into claim 3 is doomed to failure (Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at 15; Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 610). The article in question expressly concerned the commercial use of Fosamax in accordance with the then approved regime. This was the oral administration of Fosamax which was a pharmaceutically acceptable salt of alendronate selected from the group consisting of sodium, potassium, calcium, magnesium and ammonium salts at the then recommended and approved effective amount for unit dosage for the prevention and treatment of osteoporosis. The footnoted sources flesh out the context. The article discloses a continuous schedule having a dosage interval which is once weekly. Claim 3 is invalid.
106 It is submitted for Arrow that, given the recommended daily doses of 5 mg or 10 mg, it would be inevitable that the skilled reader would extrapolate to a weekly dosage of 35 mg or 70 mg thus anticipating claims 1, 2, 5, 6, 9 and 10. Whilst the reverse infringement test should not be applied literally or pedantically, I would not be prepared to read in that extrapolation for the purposes of the application of that test. Even if (contrary to the submission for Merck) the disclosure relates (inter alia) to the compositions, those claims are limited to a dosage amount but the disclosure is not.
107 It is submitted for Merck that the decision of the Full Court in Faulding establishes that to suggest that something needs to be tested is not to anticipate that which is suggested. Black CJ and Lehane J said (at [67]):
‘What all those authorities contemplate, in our view, is that a prior publication, if it is to destroy novelty, must give a direction or make a recommendation or suggestion which will result, if the skilled reader follows it, in the claimed invention.’
That statement of principle was based upon a review of certain of the authorities. Those authorities stand for the proposition that the claimed invention must be disclosed as such and not simply as a possibility. If the Lunar News article had said ‘in view of these problems a continuous dosing schedule with various intervals greater than one day should be tested’ it would not anticipate claim 3, even though a weekly dosage interval would be both technically and practically contemplated by that suggestion. On the contrary, here, the disclosure is quite precise and accords with the gist of the claimed invention. I do not accept the submission on behalf of Merck that the passage in question from Faulding adds an additional requirement for anticipation, namely that the publication should recommend the use of the invention as disclosed. That is not what the passage from Faulding says and it does not follow from the authorities analysed in that judgment. The essential difference in the treatment of the prior publications in Faulding lay in the view that one publication pointed clearly to one solution which was the invention rather than other publications which did not so point. That was a factual rather than a legal judgment and cannot be translated to the present circumstances.
108 In my opinion, the mere fact that a test of a defined solution to a problem is suggested does not avoid the disclosure being an anticipation. A drawing of a mechanical device with a caption ‘should be tested for speed’ would be an anticipation of a claim for that mechanical device. This point has particular cogency in relation to the present field. No invention can be used in the treatment of humans without extensive testing. In the present case, the patent was applied for before that testing had even commenced. The same reasoning is an answer to the argument advanced on behalf of Merck that the disclosure could not amount to an anticipation as no sensible medical practitioner would have acted upon it without adequate testing for safety. Even if that were correct (and it is contrary to some of the expert evidence led on behalf of Arrow) it would not affect the fact of disclosure having been made and being publicly available.
July 1996
109 It will be recalled that the article included the following:
‘The difficulties with oral bisphosphonates may favour their episodic (once/week) … administration. Even oral alendronate potentially could be given in a 40 or 80 mg dose once/week to avoid dosing problems and reduce costs.’
110 It is to be borne in mind, of course, that the context of the article was discussion of a method for prevention and treatment of osteoporosis by the oral administration of Fosamax. Applying the principles to which I have referred in dealing with the April 1996 edition, this is a clear anticipation of claims 1 and 2 subject to the difference between 35 mg and 40 mg and between 70 mg and 80 mg. In my opinion, those differences are immaterial. First, the claims are imprecise, being ‘about’ the identified figure. Secondly, the dosage of 40 mg is equivalent as a matter of substance to 35 mg in these circumstances and 80 mg is equivalent as a matter of substance in these circumstances to 70 mg. It is clear enough on the evidence that, in terms of both safety and efficacy, there is no relevant difference. I have referred to many internal Merck documents that demonstrate that fact. Furthermore, the examples in the patent are equivalent to 80 mg. In my view, administering a dose of 40 mg otherwise in accordance with claim 1 would infringe claim 1 and administration of 80 mg otherwise in accordance with claim 2 would infringe claim 2 (Azuko Pty Ltd v Old Digger Pty Ltd (2001) 52 IPR 75 at [35]–[45], [161]). The reverse infringement test should be applied in no less a sensible fashion. Claims 1 and 2 are invalid on this basis.
111 Claim 3 is also anticipated. The context of the article is discussion of a method of preventing or treating osteoporosis in a human; oral administration of Fosamax (which is alendronate monosodium trihydrate) is identified; the reference to 40 mg or 80 mg doses answers the description as a unit dose (however taken) and it is plain that 40 mg and 80 mg respectively are put forward as being pharmaceutically effective amounts if taken orally. The footnoted articles assist in spelling this out. Most importantly, the gist of the claim, being the weekly interval, is expressly identified.
112 The answer to the question whether the composition claims are anticipated by this publication is not so clear. The statement assumes rather than states that a dose as claimed was feasible and available. Even if the footnoted articles are taken into account, I do not think that the reference is clear enough to amount to anticipation.
April 1997
113 It will be recalled that the article includes the following:
‘In the human, there is little difference between 5 and 10 mg/day of alendronate, so the 10 mg dose theoretically could be given only 3X a week or the 40 mg dose once per week.’
114 In the context in which it appears, and for the reasons that have been explained in relation to the earlier articles, in my opinion, that disclosure anticipates claims 1 and 3. In my opinion, it does not anticipate claims 5 and 6.
Tablets
115 Arrow also relies upon the marketing by Merck or its related or affiliated companies in Australia of 10 mg and 40 mg tablets of alendronate sodium from 1 October 1996 (in each case with the packet) as being anticipation of claims 5, 6, 9 and 10.
116 In my opinion a 40 mg tablet is the equivalent for relevant purposes of a tablet that is ‘about 35 mg’. Claims 5 and 6 are therefore anticipated. Whether claims 9 and 10 are anticipated on that basis depends upon construction of those claims. In Merck & Co Inc’s Patents [2003] FSR 29 at [70], Jacob J held that the UK claim for ‘a unit dosage form which comprises about 70 mg’ was not limited to a single pill or sachet, relying upon a passage from the specification which is also included in the Australian specification. That construction was upheld in the Court of Appeal (Merck & Co Inc’s Patents [2004] FSR 16). Whilst minds may differ upon the point, it is appropriate to follow the Court of Appeal in that respect. There is thus anticipation of claims 9 and 10 as two 40 mg or seven 10 mg pills would comprise ‘about 70 mg’.
Entitlement
117 This ground is based upon s 15(1)(a) and s 138(3)(a) of the Act. The only basis which was pursued is that Mazess is the only true inventor or, alternatively, that he is one of the true inventors. If that is so, it is submitted that the claimed inventors were not entitled to the patent. The findings which I have already made concerning the provenance of the alleged invention are inconsistent with the case sought to be made. Mazess was not either the only or one of the true inventors.
False Suggestion
118 This ground is based upon s 138(3)(d) of the Act. In its final submissions Arrow relied upon two bases for this ground:
(1) That the patentee falsely suggested that the only inventors of the patent were Anastasia Daifotis, Arthur Santora II and John Yates when in fact the real inventor was Richard B Mazess. That basis fails for reasons already explained.
(2) That the specification misrepresents the teaching of the Chesnut article. This depends upon the following portion of the specification:
‘In other words, it is found that the administration of a bisphosphonate at a high relative dosage at a low relative dosing frequency causes less adverse gastrointestinal effects, particularly esophageal effects, compared to the administration of a low relative dosage at a high relative dosing frequency. This result is surprising in view of the teachings suggesting that adverse gastrointestinal effects would be expected to increase as a function of increasing bisphosphonate dosage.’
Earlier, it had been said:
‘The degree of adverse gastrointestinal effects of bisphosphonates has been shown to increase with increasing dose’
and the Chesnut article is cited.
119 It is submitted on behalf of Arrow that the Chesnut article was not represented in that way by Merck in its application to the FDA or by the inventors in the article published in 2000 by Bone et al, ‘Weekly administration of Alendronate: Rationale and Plan for Clinical Assessment’ in Vol 22 of the periodical Clinical Therapeutics at p 15. It was also submitted that the evidence of Dr Diamond and Professor Sambrook was to the contrary and that, on proper examination, the evidence of each of Professor Ebeling and Yates was inconsistent with the statement in the patent.
120 This is a difficult ground to establish where the statement that is attacked purports to be a conclusion drawn from a published article. The patent office and others with an interest are all able to read the article and form independent judgments. Establishment of the ground would at least require a conclusion that the representation was deliberately false and intended to mislead. I was not invited to make such a finding and I do not think that there is a basis for doing so. (Cf ICI Chemicals & Polymers Ltd v Lubrizol Corporation Inc at [91]).
Fair Basis
121 This ground arises from s 40(3):
‘The claim or claims must be … fairly based on the matter described in the specification.’
and s 138(3)(f).
122 The attack is upon the pharmaceutical composition claims. The patent only discloses compositions useful for carrying out the methods of treatment disclosed but it is submitted that none of the relevant claims contains any such limitation and thus they travel beyond the disclosure (Kimberley-Clark Australia Pty Ltd v Arico Trading International Pty Ltd at [14]). In my opinion, this is not a valid objection in this case. The purpose of such compositions is clearly disclosed in the specification, namely, to facilitate the suggested method.
123 An endeavour was made in final submissions to attack claim 3 upon the basis that it travelled beyond the disclosure in the specification because there was no disclosure of an increased dosing interval (reduced dosing frequency) without a corresponding increase in dosage. That ground was not particularised and Arrow is not entitled to rely upon it. No argument was addressed in relation to the other pre-trial particulars of invalidity on this ground. I should note that no issue of priority date was ultimately pressed because of the way the argument ran.
inventive step
124 In cases such as this it is often convenient for a trial judge to deal with issues which are not necessary to be decided in an endeavour to avoid a new trial in the event that different views are taken on appeal, notwithstanding that to do so involves judicial time and effort which might be wasted and means the delaying of judgment in the instant and other cases. I have followed that course in relation to a number of grounds already. I do not propose to do so in relation to the alleged lack of an inventive step, or obviousness as it is sometimes called, in relation to the method claims. To do so would involve grappling with a considerable body of evidence. That exercise would have an air of unreality. It will be apparent from what I have said in relation to invention and manner of manufacture that I do not regard the alleged invention as having subject matter. I do not regard a choice between dosage regimes of a patented drug in the circumstances of this case as involving an inventive step sufficient to found a stand-alone patent. Ingenuity might be exercised, but not invention. Furthermore, in conducting that exercise I would have to ignore all of the Lunar News material and much of that which I know about the actual provenance of the alleged invention from the internal Merck material. I should add that I did not gain any significant impression as to the credit of the relevant witnesses from any observation of demeanour as such. In addition to the affidavit evidence, there is a full transcript of the oral evidence and there is much relevant objective documentary material in evidence.
125 I can, however, deal with the composition claims. I have held that it is plain on the face of the specification that there is no inventive step involved in formulating the composition in a form appropriate for administration – whether in tablet or liquid form. Reference is only made to standard mixing and formulation techniques. I have pointed out that, even if the dosage regime is novel, that of itself does not confer novelty on the manner of administration or the substance involved. It was clearly part of common general knowledge by the priority date that tablets of up to 40 mg active ingredient of alendronate were on the market. That answers the description in claims 5 and 6 in substance. Even if one 70 mg tablet were contemplated by claims 9 and 10 (contrary to the United Kingdom decisions), there is no evidence to suggest that there is any inventive step in formulating a 70 mg tablet or the equivalent liquid.
Conclusion
126 Proposed claims 1, 2, 3, 5, 6, 9 and 10 are invalid. I will hear the parties upon the formal orders to be made. Arrow should bring in short minutes of order. As to costs, my present view is that Merck should pay Arrow’s costs of the proceeding, excluding costs relating to particulars of invalidity on the ground of novelty which were not pursued at trial. The parties should draw to my attention any omission to deal with any significant live issue in these reasons.
| I certify that the preceding one hundred and twenty-six (126) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justice Gyles. |
Associate:
Dated: 6 October 2004
| Counsel for the Applicant: | DK Catterns QC; SCG Burley; MG Small |
| | |
| Solicitor for the Applicant: | Baker & McKenzie |
| | |
| Counsel for the Respondent: | JMcL Emmerson QC; KJ Howard |
| | |
| Solicitor for the Respondent: | Cropper Parkhill |
| | |
| Date of Hearing: | 25–28, 31 May, 1–4, 8–11 June 2004 |
| | |
| Date of Judgment: | 6 October 2004 |

