
FEDERAL COURT OF AUSTRALIA
GlaxoSmithKline Consumer Healthcare Investments (Ireland) (No 2) Limited v Generic Partners Pty Limited [2018] FCAFC 71
Table of Corrections | |
Para [69] first sentence the words “VanKel Technology Group (“VanKel”) be amended to read “VanKel” | |
Para [126] the quotation be amended to read as follows: “A spacer for a building foundation form work arrangement wherein a plurality of boxes are located in spaced apart relationship to define therebetween channels, the spacer including outermost engaging surfaces adapted to engage against both sides adjoining a corner of a first box, and at the same time against both corners [sides] adjoining a corner…” | |
ORDERS
DATE OF ORDER: |
THE COURT ORDERS THAT:
2. The cross-appeal be dismissed.
3. Within 7 days the parties file and exchange written submissions (limited to 2 pages in length) in relation to the costs of the appeal and the cross-appeal.
4. Within 14 days the parties file and exchange submissions in reply (limited to 2 pages in length).
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
ORDERS
VID 934 of 2016 | ||
| ||
BETWEEN: | GLAXOSMITHKLINE CONSUMER HEALTHCARE INVESTMENTS (IRELAND) (NO. 2) LIMITED First Appellant GLAXOSMITHKLINE AUSTRALIA PTY LIMITED Second Appellant | |
AND: | APOTEX PTY LTD (096 916 148) Respondent | |
AND BETWEEN: | APOTEX PTY LTD (096 916 148) Cross-Appellant | |
AND: | GLAXOSMITHKLINE CONSUMER HEALTHCARE INVESTMENTS (IRELAND) (NO. 2) LIMITED First Cross-Respondent GLAXOSMITHKLINE AUSTRALIA PTY LTD Second Cross-Respondent | |
JUDGES: | MIDDLETON, NICHOLAS AND BURLEY JJ |
DATE OF ORDER: | 10 MAY 2018 |
THE COURT ORDERS THAT:
1. The appeal be dismissed.
2. The cross-appeal be dismissed.
3. Within 7 days the parties file and exchange written submissions (limited to 2 pages in length) in relation to the costs of the appeal and the cross-appeal.
4. Within 14 days the parties file and exchange submissions in reply (limited to 2 pages in length).
Note: Entry of orders is dealt with in Rule 39.32 of the Federal Court Rules 2011.
THE COURT:
INTRODUCTION
1 Before us are two appeals from a judgment of the primary judge dismissing proceedings brought by the appellants for infringement of claims 1 and dependent claims 2 to 6, 8 to 11, 13 and 14 (“the relevant claims”) of Australian Patent No 2001260212 (‘the Patent”) and dismissing cross-claims seeking orders for the revocation of those claims. The primary judge held that each of claim 1 and the dependent claims was valid but not infringed.
2 The invention the subject of the Patent is a pharmaceutical composition in the form of a tablet containing paracetamol in an immediate release phase and a sustained release phase. It is an object of the invention to maintain therapeutic levels of paracetamol for an extended period so as to provide pain relief for longer than conventional formulations.
3 The appellants in both proceedings (which we shall refer to as “GSK”) are the patentee and exclusive licensee of the Patent. By their notices of appeal the appellants challenge the primary judge’s non-infringement finding.
4 The respondent and cross-claimant (“Apotex”) to the first proceeding (No VID 571 of 2014) and the respondent and cross-claimant (“Generic Partners”) to the second proceeding (No VID 638 of 2014) have each filed a notice of contention and notice of cross-appeal. These challenge the primary judge’s finding that each of the relevant claims was fairly based on matter described in the complete specification of the Patent. They also contend in their cross-appeals, that if (contrary to their principal submission) the primary judge erred in dismissing the infringement case, then his Honour should have revoked the relevant claims on the ground that they did not define an invention and were not clear within the meaning of ss 40(2)(b) and 40(3) of the Patents Act 1990 (Cth) (“the Act”) as it stood at the relevant time.
5 By their cross-appeals, the respondents also challenge the primary judge’s finding that the best method known to the patent applicant of performing the invention was disclosed in the Patent as required by s 40(2)(a) of the Act.
THE PATENT
Background to the invention
6 The complete specification states (page 1 lines 3 to 23):
The present invention relates to pharmaceutical compositions containing N-acetyl-p-aminophenol, known by the generic names paracetamol, acetaminophen and APAP (hereinafter referred to as paracetamol). In particular, the invention relates to a sustained release paracetamol formulation having an advantageous pharmacokinetic profile.
Paracetamol is an analgesic and antipyretic agent which is widely used in prescription and non-prescription medicines, often in combination with other biologically active compounds.
The elimination half-life of paracetamol is reported to be in the range of 1.9 – 2.5 hours. Its absorption following oral doses of conventional immediate release tablets is characterised by passive absorption with high bioavailability (80%) and rapidly occurring maximum plasma concentration (tmax 30–90 min). These characteristics determine the conventional dosage regimen of 1000mg every 4 to 6 hours for the drug. Although this regimen is acceptable in the short-term treatment of acute pain, it becomes inconvenient in the context of long-term treatment of sub-chronic or chronic pain. Therefore, extended release paracetamol may improve patient’s quality of life by reducing the number of doses to be taken and providing steadier levels of the drug in the blood as determined by plasma or serum drug concentrations.
7 The Patent states at page 1 lines 25 to 28 that a paracetamol product designed for three times a day (t.i.d.) dosing should contain enough paracetamol (about 600mg to 667 mg per tablet) to give close to the maximum daily dose when two tablets are taken three times a day.
8 At page 1 lines 29 to 32 to page 2 line 3 the Patent refers to European Patent EP-A-305051 (“McNeil”) which is said to disclose a sustained release bilayer tablet containing 650 or 667mg of paracetamol which contains an equal amount of paracetamol in the immediate and sustained release layers in which the sustained release layer is provided by a matrix comprising a mixture of hydroxyethylcellulose and polyvinyl-pyrrolidone. Such a tablet is said to be marketed in the US by McNeil Inc as Tylenol® Extended Relief.
9 The invention is then discussed by reference to the McNeil sustained release bilayer paracetamol tablet and the advantages that the invention is said to provide over that tablet. The Patent then discusses at pages 2 to 3 the desirable pharmacokinetic properties for a sustained release paracetamol product. At page 2 lines 5 to 7 the Patent notes that a sustained release paracetamol oral dosage form designed for t.i.d dosing should also provide all the benefits of immediate release paracetamol plus a sustained action.
10 The Patent notes that one potential disadvantage of a formulation containing more than the standard dose of paracetamol (500mg) is accidental or intentional overdose. The Patent states at page 2 lines 11 to 22:
One potential disadvantage concerning a formulation containing more than the standard dose of paracetamol (500mg) is accidental or intentional overdose. In such circumstances more paracetamol will be ingested from an extended release formulation compared to a conventional immediate release formulation for any given number of unit doses such as tablets. This could have serious consequences for an overdose patient, especially if a large amount of the dose is absorbed before rescue therapy could be initiated. It would therefore be preferable if the unit dose (such as a tablet) was designed to limit the amount of paracetamol absorbed in the first few hours following dosing. An advantageous sustained release formulation should therefore demonstrate a lower mean Cmax (preferably at least 20% lower) than a conventional immediate release formulation which would be indicative of a lower initial exposure.
11 At page 2 lines 24 to 28 of the Patent it is recognised that with an oral paracetamol product designed to have a lower Cmax and slower rate of absorption, the extent of absorption may also be decreased and that this could then lead to sub-therapeutic systemic levels of drug 6 to 8 hours following dosing thus leading to premature onset of pain before administration of a further dose. At page 3 lines 1 to 3 it is said that such a product should maintain therapeutic levels of paracetamol in plasma for extended periods following dosing and hence provide analgesia for longer than a conventional immediate release tablet or capsule.
12 The Patent notes that it is desirable for plasma levels associated with a sustained release formulation to be maintained at therapeutic levels (>3mcg/ml) for longer than a conventional immediate release formulation. The Patent states at page 3 lines 7 to 21:
Whilst such a formulation should have a lower Cmax compared to a conventional immediate formulation, it is still desirable to have a fast onset of action, therefore initial levels of drug in plasma should be rapidly attained (preferably within 30 minutes) and maintained at therapeutic levels of >3mcg/ml for at least 1.3 hours and preferably 1.5 hours longer than a standard immediate release tablet or capsule. In addition the extent of absorption should be equivalent to a conventional immediate release formulation.
Furthermore, upon multiple dosing of a sustained release formulation the steady state plasma levels of paracetamol should be more constant than those achieved following multiple dosing of a conventional immediate release formulation. A convenient measure of the fluctuation in plasma concentrations is the fluctuation index (FI) which is defined as (Cmax – Cmin) / Caverage. A low FI number (ie <1) is considered to be advantageous as it suggests a reduction in the variability of plasma concentrations indicative of a safer product.
13 At page 3 line 23 to page 4 line 10 the Patent states:
In summary, an advantageous sustained release paracetamol product for oral administration should possess the following pharmacokinetic attributes:
(1) therapeutically active drug plasma concentrations should be attained rapidly.
(2) the mean maximum plasma concentration (Cmax) should be at least 20% lower compared to standard immediate release formulation;
(3) a mean plasma concentration of at least 3 mcg/ml should be maintained for at least 1.3 hours longer (preferably 1.5 hours longer) than a standard immediate release formulation;
(4) the extent of absorption should be equivalent to a conventional immediate release paracetamol;
(5) plasma levels of paracetamol following multiple dosing should be more constant compared to multiple dosing of an immediate release formulation as measured by a reduction in the fluctuation index.
14 At page 4 lines 12 to 14 the Patent states:
Surprisingly it has now been discovered that such an advantageous pharmacokinetic profile can be provided by a two phase (immediate release and sustained release) formulation of paracetamol which satisfied a unique in vitro dissolution profile.
15 The Patent contains two consistory clauses. At page 4 line 27 to page 4a line 20 the Patent states:
In one aspect, the present invention provides a pharmaceutical composition comprising a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol,
the immediate release phase being in one layer and comprising from about 10 to 45% by weight of the total paracetamol; and
the sustained release phase being in the other layer and comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof;
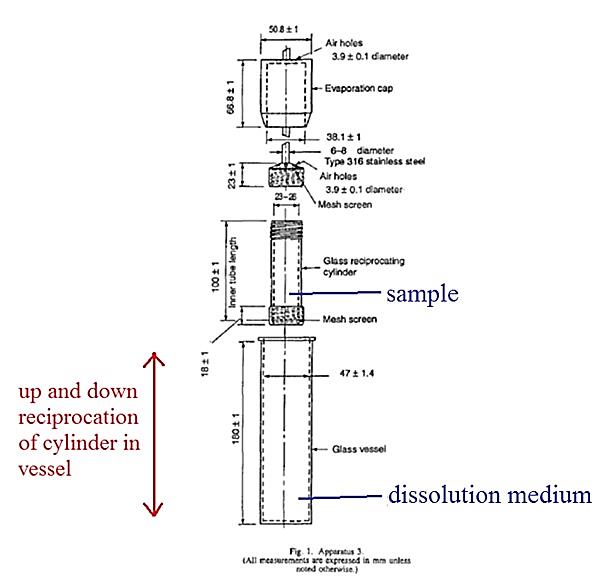
said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier, wherein said composition has an in vitro paracetamol dissolution profile (as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/min) with the following constraints:
• 30 to 48% released after 15 minutes
• 56 to 75% released after 60 minutes
• >85% released after 180 minutes.
Accordingly, in another aspect the present invention provides a pharmaceutical composition, having an immediate release phase and a sustained release phase of paracetamol, said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier, characterised in having an in vitro paracetamol dissolution profile (as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HC1 at 37C set at a cycle speed of strokes/min) with the following constraints:
• 30 to 48% released after 15 minutes
• 56 to 75% released after 60 minutes
• >85% released after 180 minutes.
16 Each of the consistory clauses refers to “an in vitro paracetamol dissolution profile as determined by USP type III apparatus, reciprocating basket …”. These words are also used in claim 1.
17 In essence, GSK contends that the person skilled in the art (“PSA”) would have understood these words as referring to a “USP type III apparatus, reciprocating cylinder” and that his Honour erred by declining to interpret them in that way.
The detailed description of the invention
18 At page 5 lines 15 to 32 of the Patent it is said:
The immediate release phase and the sustained release phase both contain paracetamol and a pharmaceutically acceptable carrier and are suitably combined together into a unit dose form. For example the immediate release phase and the sustained release phase can be separate blends, granules or pellets which can be mixed together before being compressed into a tablet or being filled into a capsule. A preferred unit dose form is a bilayer tablet having an immediate release layer of paracetamol and a sustained release layer of paracetamol.
Suitably the sustained release phase comprises a matrix-forming polymer to provide a sustained release of paracetamol.
Examples of matrix-forming polymers include both water soluble and water insoluble polymers or mixtures thereof, with soluble polymers being preferred. Examples of water soluble polymers include hydroxypropylmethylcellulose, hydroxyethylcellulose, carboxymethylcellulose, sodium carboxymethylcellulose, methacrylate hydrogels, polyethylene glycols and xanthan gum. An example of a water insoluble polymer is ethylcellulose. A preferred matrix-forming polymer is hydroxypropylmethylcellulose.
19 At page 6 lines 2 to 9 of the Patent it is said:
The amount of matrix-forming polymer in the sustained release phase and the relative amounts of paracetamol in the sustained release and immediate release phases are selected so as to provide the desired in vitro dissolution rate as herein before described.
Thus, the matrix-forming polymer is suitably present in an amount from 0.5 to 10%, preferably from 1 to 6%, and more preferably from 2 to 4% by weight of the sustained release phase.
20 At page 6 lines 20 to 28 of the Patent it is said:
Compositions of the present invention will generally contain at least one pharmaceutically acceptable carrier conventionally used in the art of tablet and/or capsule formulation. Suitable carriers which may be incorporated include lubricants, for example magnesium stearate and stearic acid; disintegrants, for example cellulose derivatives and starches; binders, for example modified starches, cellulose derivatives and polyvinylpyrrolidone; glidants, for example colloidol silicas; compression aids, for example cellulose derivatives; as well as preservatives, suspending agents, wetting agents, flavouring agents, bulking agents, adhesives, colouring agents, sweetening agents appropriate to their form.
Example 1
21 The invention is then illustrated by way of four example formulations (Formulations A to D), two of which are the subject of Example 1 and said to have an in vitro dissolution profile falling outside the scope of the invention (Formulations A and B). The other two formulations are said to have an in vitro dissolution profile falling within the scope of the invention (Formulations C and D).
22 The ingredients of Formulations A and B are listed in the table on page 9 of the Patent. The paracetamol used in the immediate release layer of Formulations A and B is identified as DC90. The Patent notes at page 9 lines 30 to 32 that this is a commercially available directly compressible paracetamol granulation containing about 90% by weight of paracetamol together with pregelatinised starch, croscarmellose sodium, polyvinylpyrrolidone and stearic acid.
23 Formulations A and B were assessed in a pharmacokinetic study in healthy fasted volunteers using 500mg immediate release paracetamol tablets as a control (see page 10 lines 10 to 14). The results are presented graphically in Figure 1 on page 11. The Patent notes at page 11 lines 5 to 7 that neither Formulation A nor B met the criterion of achieving a mean paracetamol plasma concentration of 3mcg/ml for at least 1.5 hours longer than the immediate release tablet.
24 The dissolution profile for each of the formulations the subject of Example 1 and each of the other formulations the subject of Examples 2 and 3 (discussed below) were all characterised using an apparatus described at page 10 lines 1 to 3, page 12 lines 15 to 17 and page 15 lines 20 to 22 as “the USP type III apparatus (reciprocating basket)”.
Example 2
25 Example 2 compares the properties of a commercially available immediate release 500mg paracetamol tablet with a sustained release bilayer tablet having an in vitro dissolution profile falling within the scope of the invention (Formulation C). The ingredients for Formulation C are set out in the table on page 12 of the Patent. The ingredients listed include “High viscosity HPMC” (HPMC refers to hydroxypropylmethylcellulose) which acts as the matrix forming polymer. However, the complete specification does not specify any particular grade or viscosity of the “high viscosity HPMC” that is referred to in the list of ingredients for Formulation C. The dissolution profile for Formulation C is set out in the table at page 13.
26 Formulation C was assessed in a pharmacokinetic study using a panel of 26 healthy volunteers and a two tablet dose of a standard immediate release 500mg paracetamol tablet comparator. Formulation C was said to have met all of the relevant pharmacokinetic criteria for an ideal sustained release paracetamol tablet. In particular, in comparison to the immediate release product, Formulation C was found to be bioequivalent with respect to the area under the curve (“AUC”), and with a significantly lower Cmax. Mean plasma levels for Formulation C remained above the therapeutic level of 3mcg/ml for longer than for the immediate release product.
Example 3
27 Example 3 compares the properties of a commercially available immediate release 500mg paracetamol tablet with Formulation D, which is described as a sustained release bilayer tablet having an in vitro dissolution profile falling within the scope of the invention.
28 The bilayer tablet of Formulation D is said to be “essentially similar to Formulation C but contained a total of 665mg of paracetamol and had a slightly different ratio of sustained release to immediate release paracetamol” (Patent page 15 lines 15 to 18). No additional information is provided in relation to the grade or viscosity of the high viscosity HPMC used beyond what appears in the table on page 12. The dissolution profile for Formulation D appears in a table on page 15 of the Patent.
29 The Patent states that the two treatments were shown to be bioequivalent with respect to AUC24–48 and that Formulation D provided a lower Cmax, a higher Cmin and a substantially lower fluctuation index (FI) compared to the immediate release formulation. At page 3 lines 15 to 21 an advantage of a low FI number is described:
Furthermore, upon multiple dosing of a sustained release formulation the steady state plasma levels of paracetamol should be more constant than those achieved following multiple dosing of a conventional immediate release formulation. A convenient measure of the fluctuation in plasma concentrations is the fluctuation index (FI) which is defined as (Cmax – Cmin) / Caverage. A low FI number (ie < 1) is considered to be advantageous as it suggests a reduction in the variability of plasma concentrations indicative of a safer product.
30 At page 17 lines 7 to 13 the Patent states that Tylenol Extended Relief does not have a lower FI than the immediate release tablet:
The substantially lower FI for the SR [ie. sustained release] product is surprising considering previous reports for a steady state biostudy conducted with a 650mg bilayer tablet (Tylenol Extended Relief) which showed that the SR product had a numerically higher FI (of 1.49) compared to a reference 500mg IR tablet (of 1.44) as illustrated in Figure 4. Furthermore, Paracetamol plasma levels were maintained substantially above 3mcg/ml for the entire study period, which is in contrast to the steady state study reported for Tylenol® Extended relief.
31 The low FI number of <1 found for Formulation D is said at page 18 lines 5 to 7 to be “particularly advantageous” for a sustained release formulation as it indicates a reduction in the variability of plasma concentration suggesting a much safer and more reliable product.
32 Figure 4 on page 18 compares the FI of Tylenol Extended Relief and an immediate release tablet based on a steady state biostudy of the two. It shows that the Tylenol Extended Relief had a numerically higher FI than the immediate release formulation. Figure 4 shows that sustained release formulations do not, as a matter of course, have a lower FI compared to a corresponding immediate release formulation. Figure 4 also illustrates that the plasma level of Tylenol Extended Relief fell below 3mcg/ml on a number of occasions.
Example 4
33 Example 4 compares the clinical properties of a commercially available paracetamol 500mg immediate release tablet with a sustained release bilayer tablet of Formulation D based on a multicentre, single dose, double-blind study. The Patent reports the results of the study at page 20 lines 6 to 25 and states:
Based on the patient global assessment at 4 hours, the extended release product was shown to be equivalent or better than the immediate release product. A successful response was defined as a ‘very good’ or ‘excellent’ rating: 88 of 252 (35.1%) patients treated with the SR paracetamol formulation gave a successful response compared with 71 of 258 (27.7%) patients treated with standard IR paracetamol. Equivalence was concluded from the 90% confidence interval of the treatment difference (7.3% in favour of SR paracetamol) between the two formulations.
There was no significant difference between SR paracetamol and standard IR paracetamol in either development of analgesia (time to peak pain relief, time to peak pain intensity difference, total pain relief 1 hour after treatment) or peak analgesic effect (peak pain relief, peak pain intensity difference). However, the SR tablet was significantly more effective than standard IR paracetamol for the summed pain analogue intensity difference at 6 hours (p = 0.0344) and 8 hours (p = 0.0500). Furthermore, the median time to re-medication was longer for SR paracetamol (245 mins) compared with standard IR paracetamol (190 mins). Although this was not statistically significant, it was clear from the separation of the two curves on the Kaplan-Meier plot that a smaller proportion of patients treated with SR paracetamol re-medicated between approximately 3 and 6 hours compared with standard IR paracetamol.
34 The Patent concludes at page 20 lines 27 to 30 that the Example 4 results indicate that the sustained release tablet gave rapid analgesia which was maintained for up to eight hours following dosing and the sustained release tablet had a longer duration of action than the immediate release paracetamol.
The relevant claims
35 The Patent claims pharmaceutical compositions containing paracetamol in the form of “a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol”. It was common ground that the priority date for each of the relevant claims was 19 April 2000.
36 Claim 1 is for:
A pharmaceutical composition comprising
a bilayer tablet having an immediate release phase of paracetamol and a sustained release phase of paracetamol,
the immediate release phase being in one layer and comprising from about 10 to 45% by weight of the total paracetamol; and
the sustained release phase being in the other layer and comprising from about 55% to 90% by weight of the total paracetamol in admixture with a matrix forming polymer or a mixture thereof;
said composition comprising from 600 to 700mg of paracetamol per unit dose and a pharmaceutically acceptable carrier,
wherein said composition has an in vitro paracetamol dissolution profile (as determined by the USP type III apparatus, reciprocating basket, with 250ml of 0.1M HCl at 37C set at a cycle speed of 15 strokes/min) with the following constraints:
• 30 to 48% released after 15 minutes
• 56 to 75% released after 60 minutes
• >85% released after 180 minutes.
37 All of the other relevant claims are dependent on claim 1. Claims 2 to 3 specify narrower in vitro dissolution profiles for the claimed composition than does claim 1. Claims 4 to 6 specify narrower ranges of paracetamol per unit dose than does claim 1. Claims 6 to 11 include additional requirements to the characteristics of the matrix forming polymer referred to in claim 1. And claims 13 and 14 impose additional requirements with respect to the proportion of paracetamol in the sustained release component of the claimed composition relative to the immediate release component.
The PRIMARY JUDGE’S REASONS
The skilled addressee
38 An important step that must be performed in resolving any question of patent construction is to ascertain the identity of the skilled addressee or, in the language used in s 7 of the Act, the person skilled in the art. The primary judge dealt with this question comprehensively and in a manner that neither party suggested involved any error. His Honour found at [280] that the Patent is addressed essentially to a pharmaceutical formulator who is a member of a wider team that would include people with expertise in medicinal chemistry, analytical chemistry, pharmacokinetics and pharmaceutical formulation development. The primary judge also found at [280] that the formulator would work with, and seek guidance from, a scientist with particular expertise in the dissolution testing of extended release oral dosage forms using USP apparatus, in particular the USP type III, (which we discuss below) to carry out the claimed invention.
The common general knowledge
39 There was a debate before the primary judge as to whether the Patent was to be construed in light of the common general knowledge as at the date of the filing of the complete specification or as at the priority date. The primary judge accepted GSK’s submission that the priority date was the date for assessing the common general knowledge for the purpose of construing the Patent. His Honour’s conclusion on this issue was not challenged in the appeal and we need not say anything more about it.
40 The primary judge made a number of findings as to the state of the common general knowledge as at the priority date. We shall summarise those findings that are relevant to the determination of the appeal and cross-appeal.
41 Extended release (“ER”) or sustained release (“SR”) pharmaceutical compositions suitable for oral administration were known well before the priority date. A sustained release pharmaceutical composition in which paracetamol is the active pharmaceutical ingredient should be non-toxic and provide pain relief over a longer period than an immediate release formulation. This is achieved by including in the formulation larger quantities of paracetamol in a single dose that are equivalent to multiple doses of an immediate release formulation. The drug is released so as to achieve a minimum effective plasma concentration (“MEC”) of the drug over an extended period of time while also avoiding unintended side effects that may occur if the concentration were to exceed the minimum toxic plasma concentration (“MTC”).
Dissolution Profile
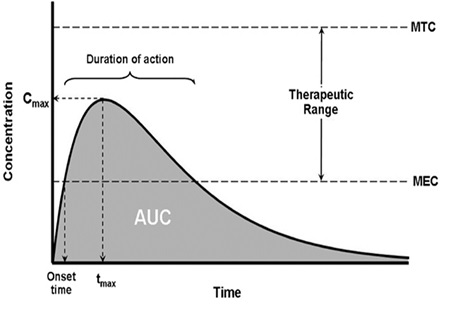
42 When drug plasma concentration (y-axis) versus time (x-axis) is plotted graphically, the area under the plotted curve (the curve) represents the plasma concentration over time. The shape of the curve reflects what is referred to as the “dissolution profile” of the drug.
43 AUC represents the total exposure to the drug over time. The maximum drug plasma concentration (Cmax) and the minimum drug plasma concentration (Cmin) over a given period of time will also be apparent from the graph. The time at which Cmax is achieved is known as the tmax. Horizontal lines extending from the y-axis representing the MEC and the MTC define the area referred to as the “therapeutic window” or “therapeutic range” in which drug plasma concentrations are above the MTC and below the MEC.
Techniques for achieving sustained release
44 With a sustained release drug formulation, the drug plasma concentration should remain within the therapeutic range over an extended period. One way in which this may be achieved is through the use a of bilayer tablet having an immediate release layer and extended release layer. The immediate release layer is designed to dissolve relatively quickly whereas the sustained release layer is designed to dissolve more slowly.
45 Matrices are another way of controlling the rate of release of a drug in a solid oral dosage form such as a tablet. In a matrix system, the drug is dispersed through other materials such as a polymer or lipid with their own dissolution characteristics, which may be used to control the release of the drug and help shape its dissolution profile.
Dissolution testing
46 The rate at which a pharmaceutical formulation dissolves under certain conditions is identified by “dissolution testing”. Dissolution testing is typically carried out in conditions which are designed to mimic the dissolution of the drug in the body. Dissolution testing of pharmaceutical formulations is typically carried out in vitro under standard conditions. Standardised pharmaceutical dissolution testing apparatus are described in the United States Pharmacopeia (“USP”), a compendium published annually by the United States Pharmacopeial Convention.
47 As at the priority date, the USP 24 NF 19 was the applicable standard for dissolution testing from 1 January 2000 through to about the end of 2001. It specifies seven apparatus for testing drug dissolution from a formulation. Four of these apparatus are designed primarily for oral solid dosage forms, being USP type I, II, III and IV. The remaining three apparatus are designed primarily for drug release testing of transdermal delivery systems, suppositories and topical products. The methods and apparatus described in the USP are frequently referred to as “compendial” in the sense that they meet the requirements of the compendium.
48 USP type I apparatus is generally referred to by pharmaceutical scientists as a “basket”, USP type II apparatus as a “paddle”, USP type III apparatus as a “reciprocating cylinder” and USP type IV apparatus as a “flow through cell”. USP type I apparatus and USP type II apparatus were first included in the USP in the 1970s. USP type III apparatus was first included in the early 1990s.
49 Each of the four dissolution methods described in USP type I, II, III and IV apparatus provides different hydrodynamics. The differences in hydrodynamics have an impact on the dissolution results obtained. For example, if the same tablet is tested, the dissolution results from a test using the rotation of the paddle in USP type II apparatus will not be the same as the results when the rotating basket in USP type I apparatus is used. This is because each system creates a different fluid flow environment.
50 There is no established correlation between dissolution results measured using the different USP apparatus. Sometimes small differences are measured, other times the measured differences are large. Differences in measured dissolution profiles as between each of USP type I, II and III apparatus may not be predictable.
51 In a repeated dissolution study using any one of USP type I, II or III apparatus, variability in the measured dissolution profile for each sample of up to ±10% is considered normal and to be expected due to inherent analytical variability, and apparatus and sampling variability, although variability of ±5% is more ideal. It may be necessary to test at least 6 or 12 dosage units in a study using a particular USP apparatus, with well-defined and consistent testing parameters, in order to determine reproducibility with statistical reliability. The USP specifies the number of dosage units to test, which ranges from an initial 6 dosage units up to 24 dosage units.
52 Prior to undertaking a dissolution study, each USP apparatus has to be calibrated. Calibration involves making sure that all parts of the apparatus are in alignment and the equipment is functional with no mechanical failures. Reference standards can be purchased from the USP for calibration of a dissolution apparatus. These reference standards include disintegrating tablets, non-disintegrating tablets, extended-release tablets and extended-release beads, each of which were available prior to the priority date. The USP calibration standard specifies all parameters for the dissolution test as well as specific dissolution ranges for each measured time point. If the results fall within the dissolution ranges specified for the calibrator reference dosage form provided by the USP, it will indicate that the dissolution apparatus has been appropriately calibrated.
53 In some circumstances, the FDA may waive the requirement for in vivo bioavailability studies that it might otherwise require before granting a product approval for a pharmaceutical product. This is what is known as a “biowaiver”. The FDA provides guidance to guide pharmaceutical companies as to the dissolution testing required to obtain a biowaiver. The FDA requires highly reliable, reproducible and discriminatory dissolution testing rather than mere adherence to the USP.
54 In assessing the parameters of the dissolution method to be used to create a set of reference data, it is sometimes necessary to modify the standard USP dissolution apparatus in order to achieve reproducible results. Modifications to the apparatus to achieve the required reproducibility of results may be made, based on the experience of individual scientists and the facilities of the laboratory in which they work, but it is important that anyone seeking to determine whether a sample dosage form has the required dissolution profile uses the same apparatus and testing conditions.
USP type I apparatus
55 USP type I apparatus uses a basket. USP type I apparatus is used for immediate-release, delayed-release and sustained-release solid dosage forms. It is comprised of a cylindrical mesh basket attached to a shaft that is placed inside a one litre round-bottom cylindrical vessel containing the dissolution medium. The dosage form being tested is placed inside the cylindrical basket which rotates inside the dissolution vessel. The speed at which the basket rotates (measured in rotations per minute) has an impact on the measured dissolution of the sample.
56 The USP specifies that the basket is made of stainless steel mesh. Different grades of mesh sizes are available. The USP specifies that 40 grade mesh be used unless otherwise specified in an applicable individual monograph.
57 The “grade” of mesh refers to the width of wire and of the openings. For example, a 40 grade mesh is specified in the USP to be a 40 x 40 mesh with 0.25mm wire diameter with wire openings of 0.40 ± 0.04mm. Where a 20 grade mesh is specified, a 20 x 20 mesh, 0.40mm wire diameter with wire openings of 0.90 ± 0.09mm is to be used.
58 The rotation speed (rpm) and grade of mesh used for the basket directly influences the hydrodynamics of the system and has a resulting impact on the rate of dissolution measured. The tighter the mesh, the more resistance there is to the dissolution medium passing through it. The 40 grade mesh is tighter than the 20 grade mesh and has a greater resistance to the passage of dissolution medium. The mesh grade used is important for reproducibility. Depending on the dosage form being tested, there may be a need to vary the mesh grade for a specific reason. For example, if the dosage form being tested included matrix-forming polymers that swell and gel (forming a gel layer around the tablet) in the dissolution medium, one would select a more open grade of mesh (e.g. 20 mesh) because the tablet is less likely to stick to a mesh with wider openings compared to a finer mesh, and so less variation should occur in the measured dissolution values.
USP type II apparatus
59 USP type II apparatus uses a paddle. USP type II apparatus is comprised of a one litre round-bottom vessel containing the dissolution medium and a paddle attached to a shaft. The sample dosage form under test is added to the vessel and allowed to sink to the bottom, below the paddle. The paddle then rotates and stirs the dissolution fluid in the vessel.
60 USP type II apparatus is used for immediate-release, delayed-release and sustained-release solid dosage forms.
USP type III apparatus
61 The USP type III apparatus was first included in the relevant standards in 1991, as an alternative to the basket (USP type I) and paddle (USP type II) apparatus for drug release testing.
62 The USP type III apparatus assembly consists of a set of cylindrical flat-bottomed glass vessels; a set of glass reciprocating cylinders; stainless steel fittings, and screens (made of suitable nonsorbing and nonreactive material) designed to fit in the bottom or top of the reciprocating cylinders; a motor and drive assembly to reciprocate or dip the cylinders vertically inside the vessels, and if desired, index the reciprocating cylinders horizontally to a different row of vessels; and a water bath which can be heated to the desired temperature.
63 In the USP type III apparatus, mesh is inserted into the bottom cap and, optionally, the top cap of the reciprocating cylinder. The purpose of the mesh is to allow adequate distribution of medium (and dissolved sample) into and out of the reciprocating cylinder, whilst retaining undissolved particles/dosage form within the reciprocating cylinder.
64 The USP type III apparatus is depicted below. This image is taken from the USP with annotations added by Professor Dressman, an expert witness called by GSK:
65 The appropriate mesh size to use to carry out the dissolution testing using the USP type III is not specified in the USP. The protocol is silent on the mesh size to be used for the USP type III apparatus. However, selecting the mesh size to carry out testing using the USP type III apparatus was a matter of routine as at the priority date for those who had a knowledge of dissolution testing including using the USP type III apparatus.
66 As at the priority date the USP type III apparatus was attractive for the study of controlled release formulations, particularly since changing the media to simulate passage through the GI tract could be easily and reproducibly achieved if required. In this regard, the primary judge referred at [191] of his reasons to Borst, I, Ugwu, S and Beckett, AH “New and Extended Applications for USP Drug Release Apparatus 3” Dissolution Technologies (February 1997) 11 who conclude at 15:
USP 3 should be considered the first line apparatus in product development of controlled release preparations, because of its usefulness and convenience in exposing products to mechanical as well as a variety of physicochemical conditions which may influence the release of products in the GI tract.
67 The USP 24 NF 19 Official Monographs for acetaminophen (paracetamol) reveal that the dissolution test indicated for acetaminophen tablets does not use the USP type III apparatus. There is no guidance for mesh size in using the USP type III apparatus.
The VanKel Documents
68 The primary judge admitted into evidence the three documents (“the VanKel documents”) which were tendered by Apotex. These were a 1999 Buyers Guide produced by VanKel Technology Group (“VanKel”); an Ordering Book from VanKel (undated but bearing a 2001 copyright statement); and certain pages from a document entitled “Dissolution Testing and Analysis: Dissolution Systems Source Book 2014-2015”.
69 The primary judge accepted that VanKel was a well-known manufacturer of USP apparatus well before 2000. However, his Honour was not satisfied that any of the VanKel documents formed part of the common general knowledge as at the priority date. This finding was not challenged by Apotex.
70 The primary judge found at [312] that the dimensions shown in the USP show that a USP type I apparatus basket would readily fit into a USP type III apparatus vessel and that this would have been known to the skilled addressee. However, it does not follow merely because a basket can fit inside the glass vessel that the modified apparatus would still work. The basket would need to be connected to the drive shaft by some means so that the basket can be pushed up and down (ie. in a reciprocating movement) inside the glass vessel. Some form of adapter would be required to make that connection.
71 The primary judge said at [312]-[313]:
[312] Apotex has asserted that a basket as used with a USP type I apparatus would fit within a USP type III apparatus vessel and that this would have been known to the skilled addressee at the relevant time. I accept, as Apotex asserts, that the dimensions shown in the USP confirm that a USP type I apparatus basket would readily fit into a USP type III apparatus vessel and that this would have been known to the skilled addressee. This is because:
(a) the outer diameter of the basket is 25.0mm ± 0.02mm;
(b) the inner diameter of the USP type III apparatus vessel is 47 ± 1.4mm (and the outer diameter of a reciprocating cylinder is itself wider than a basket at 26mm);
(c) the length of the basket is 37 ± 3mm; and
(d) the length of the USP type III apparatus vessel is 180 ± 1mm (and the length of a reciprocating cylinder is longer than a basket at 100 ± 1mm).
[313] Apotex asserts that the “basket adapter” product in the VanKel documents (I will deal with these in a moment) enabled a USP type I apparatus basket to be attached to the shaft of the USP type III apparatus. Apotex contends that the commercial availability of such a part, even if only available after the priority date, is evidence that supports the likelihood of a skilled addressee understanding such an arrangement to be practicable as at the priority date. Apotex also submits that the evidence shows that a suitable adapter was available at the priority date. For present purposes it is sufficient to say that I accept that it was part of common general knowledge as at the priority date that it may have been possible and potentially practicable to adapt USP type III apparatus to fit a basket (of the type used in USP type I apparatus) in substitute for a cylinder, but that as at the priority date it was not known that this had been done. I reject the assertion that it was common general knowledge that such an adapter was available at the priority date but I accept that it would have been known that one could have been made.
(original emphasis)
72 Since his Honour was not satisfied that the VanKel documents were common general knowledge, the question that then arose was what use if any might be made of them. His Honour said at [342]:
… In my view they can only be relevant at most as providing some flimsy evidence that as at the priority date VanKel as a manufacturer offered a relevant basket modification to the USP type III apparatus and that it was representing to the reader that this was in its view a useful modification. At best that slightly enhances the probability that I can infer that a skilled addressee as at the priority date might also not have thought such a modification to be ridiculous, even if they were not aware of such documents or what VanKel was offering. Such documents can indirectly form part of a matrix which supports the drawing of such an inference …
(original emphasis)
Additional Findings
73 There are a number of additional findings made by the primary judge in relation to the common general knowledge to which we should refer. In particular, his Honour accepted that the skilled addressee would approach the construction of “the claim 1 dissolution test” as at the priority date with an awareness of each of the following matters:
First, there is a range of standard testing apparatus specified in the USP from which a selection can be made and variables chosen in order to have an appropriate test for a specific formulation;
Second, modifications can be made to standardised apparatus in order to obtain a useful dissolution test in the sense of being reproducible and having good discrimination. However, it is important to accurately specify the equipment for use in a dissolution test so that the reader can accurately reproduce the test;
Third, the word “basket” has a clear and well understood meaning in relation to USP dissolution testing, namely, a part of the USP type I apparatus;
Fourth, the USP type III apparatus is an appropriate apparatus to use for testing of sustained release dosage forms comprising a matrix forming polymer. The USP type I apparatus (basket) and the USP type II apparatus (paddle) were known to have problems with floating or sinking oral dosage forms;
Fifth, the FDA was “resistant” to the use of dissolution testing using modified non-compendial testing apparatus by companies seeking registration of dosage forms;
Sixth, it was known that modifications to compendial USP apparatuses were developed to overcome known issues encountered when testing formulations using USP type I apparatus or USP type II apparatus and only accepted by the FDA if the modified method was shown to be “superior to other established methods”;
Seventh, when modifications to compendial testing methods were made by those conducting dissolution tests, detailed descriptions of the modifications were included in the scientific literature to enable the modified dissolution testing to be reproduced by others. The existence of such modifications would be highlighted by, for example, use of the words “modified” or “non-compendial”;
Eighth, there would be no good scientific or technical reason to use a basket instead of a cylinder with a USP type III apparatus; and
Ninth, the features of the dissolution test specified in the Patent (i.e. stroke rate, volume, temperature and dissolution medium) were consistent with what a person skilled in the art would expect for the USP type III apparatus.
74 There were two other findings sought by Apotex that the primary judge declined to make; first, that it would be sensible to attach a basket to a USP type III apparatus, for example, to simulate the more aggressive conditions of the stomach or to avoid any surface gel layer on the matrix tablet from adhering to the glass cylinder; and second, that it would be practical to attach a basket of the type specified in the USP for the USP type I apparatus to a USP type III apparatus so that the reciprocating part is a basket instead of a cylinder. As to these two matters the primary judge said at [345]:
… I am only prepared to conclude that in light of common general knowledge, a skilled addressee would have seen these as theoretical possibilities that they would not have rejected out of hand.
75 However, his Honour also recorded that none of the expert witnesses were aware as at the priority date of a modification of the USP type III apparatus or the possibility of a basket being fitted to the USP type III apparatus.
The proper construction of claim 1
76 The primary judge found at [285] that the reference in claim 1 to a “basket” would have been understood by the skilled addressee as an error and at [353] that “… the skilled addressee would read claim 1 as referring to the compendial USP type III apparatus with reciprocating cylinder, understanding the reference to ‘basket’ as being an error.”
77 His Honour then turned to the question of how he should construe the claim and, in particular, whether he should interpret “basket” to mean “cylinder”. His Honour declined to interpret “basket” to mean “cylinder” because he considered that this would be tantamount to “re-writing the claim under the guise of construction.” In support of this conclusion his Honour made the following points at [383]-[388]: the relevant integer was not ambiguous or uncertain; there was no inconsistency between the body of the specification and the claim; if “basket” means “basket” the invention still works; this was not a case of an erroneous stipulation of an underlying scientific theory upon which the invention proceeds; a patent was not to be construed with the same latitude as might be appropriate when construing a commercial contract; and it was not appropriate to adopt a method of construction that gives the patentee what it might have wished or intended to claim as opposed to what it has actually claimed.
78 His Honour’s reasons also include a consideration of various authorities relied upon by GSK including Product Management Group Pty Ltd v Blue Gentian LLC (2015) 116 IPR 54 at [111] (Kenny and Beach JJ, Nicholas J dissenting on the construction case), Adhesives Pty Ltd v Aktieselskabet Dansk Gaerings-Industri (1935) 55 CLR 523 at 545-546 per Evatt J and CCOM Pty Ltd v Jiejing Pty Ltd (1994) 51 FCR 260 at 282 and the decision of Judge Birss QC (as he then was) sitting as a judge of the Patents County Court in Lizzanno Partitions (UK) Limited v Interiors Manufacturing Limited [2013] EWPCC 12. His Honour distinguished each of these cases on the ground that they were either concerned with an error appearing in the body of the specification or, where the error appeared in a claim, it reflected an erroneous statement of theory such as, for example, that a manufacturing process needed to be “free from carbon”: see Z Electric Lamp Manufacturing Company Ld v Marples, Leach & Co Ld [1910] 27 RPC 737 at 746 per Flecher Mouton LJ and Adhesives Pty Ltd v Aktieselskabet Dansk Gaerings-Industri (1935) 55 CLR 523 at 546 per Evatt J.
79 The primary judge referred at [391] of his reasons to Bodkin, C “Patent Law in Australia” (Thomson Reuters, 2nd ed) at para [5380] in which the learned author suggests that “it is permissible for an obvious error in a claim to be corrected when construing the claim”. In support of this statement the learned author cites the decision of Gummow J in Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479 at 488.
80 The primary judge also referred at [393] to Johnson, P, Roughton, A and Cook, T “The Modern Law of Patents” (LexisNexis, 3rd ed) which states at para [6.49]:
“Where a patent claim includes a “mistake”, and this is clear to the skilled reader, then it can be ignored. This might include the addition of a word, or simply using the wrong word. Again the touchstone is whether the skilled person would understand the word to be a mistake.”
The learned authors also cite Lizzanno Partitions (UK) Limited v Interiors Manufacturing Limited [2013] EWPCC 12 at [46].
81 The primary judge did not think the authorities referenced in either text supported the propositions for which they were cited. In his Honour’s view, GSK was inviting the court to “re-write the claim under the guise of construction” which he was not able to do.
THE GROUNDS OF APPEAL
82 GSK’s principal contention was that the primary judge should have found that claim 1 refers to the compendial USP type III apparatus with a reciprocating cylinder. It submitted that, having found that the reference to “basket” would be understood by the skilled addressee to be an error, his Honour should have given effect to this finding and interpreted the claim as referring to the compendial USP type III apparatus with a reciprocating cylinder.
83 GSK contended that the primary judge erred in holding that to construe claim 1 in the manner which the skilled addressee would interpret it, would require his Honour to “re-write the claim”.
84 GSK also challenged a number of specific findings made by the primary judge. In particular, GSK submitted that his Honour erred in finding:
if “basket” means “basket”, the invention in claim 1 still works;
it would have been known that a non-compendial USP type III apparatus with a basket could have been made; and
a skilled addressee, at the priority date, would have known that a USP type I apparatus basket would readily fit into a USP type III vessel.
CHALLENGE TO FACTUAL FINDINGS
85 It is convenient to consider GSK’s challenge to his Honour’s factual findings before addressing its principal contention.
Does the invention still work?
86 The primary judge said at [385] of his reasons that if “basket” means “basket” the invention still works. There are a number of points to make about this finding.
87 The first point to make is that the primary judge accepted, correctly in our view, that in the context of describing a USP type III apparatus, a skilled addressee would understand that the word “basket” refers to a basket of the kind referred to in the description of the USP type I apparatus in the USP. In its written submissions GSK said that “[i]t is wrong to seek to construe a claim as to what the word may mean in common parlance or in non-relevant fields of industry.” We do not accept that his Honour made any such error. We reject GSK’s submission on this point.
88 GSK submitted that there was no relevant evidence that the primary judge’s construction (ie. basket means basket) would produce a workable result. This submission raises an issue of fact that does not depend on the state of the common general knowledge.
89 The fact that the specification does not direct the skilled addressee to use an adapter or teach some other means of fitting a basket to the USP III type apparatus is relevant to the sufficiency of the description of the invention, but not to the question whether it would be possible to make a non-compendial USP III type apparatus with a basket fitted to it.
90 The VanKel documents refer to a “Dissolution Basket Adapter for BIO-DIS” (“the VanKel adapter”) that appears to permit a basket to be fitted to the BIO-DIS tester (VanKel’s USP type III apparatus) in place of the cylinder. The VanKel documents therefore provided some evidence from which it can be inferred that a suitable adapter could be made. We therefore consider that the finding that the primary judge’s construction would produce a workable result was open.
91 In any event, even if we are wrong about that, it is clear that the evidence did not establish that the invention would not work if “basket means basket”. So it cannot be suggested that the primary judge’s construction should be rejected because the claimed invention will not work: cf. Martin v Scribal Pty Ltd (1954) 92 CLR 17 per Taylor J at 97; Welsh Perrin & Co Pty Ltd v Worrell (1961) 106 CLR 588 per Menzies J at 602.
Did the skilled addressee know that a non-compendial USP type III apparatus could be made?
92 The primary judge said at [313] that it was common general knowledge at the priority date that it may have been possible to adapt the USP type III apparatus to fit a basket in place of a cylinder even though it was not common general knowledge that this had been done by VanKel. Thus, the primary judge found that the skilled addressee at the priority date would appreciate that it may be possible to adapt the USP type III apparatus so that it could receive a basket in place of a cylinder. This finding was open and one that was potentially relevant to what the skilled addressee would make of the reference to “the USP type III apparatus, reciprocating basket” which is in both the body of the specification and in the claim 1.
93 It was put to Professor Dressman in cross-examination that it may have been possible as at the priority date to adapt the USP type III apparatus to fit a basket of the type used in the USP type I apparatus. Professor Dressman said that this “might or might not work”. Although his Honour was not satisfied that the skilled addressee would have known that an adapter was available at the priority date (because neither the VanKel adapter nor the VanKel documents were common general knowledge) his Honour was satisfied that the skilled addressee would have appreciated that an adapter could be made.
94 It is important to note that Professor Dressman did not give evidence to the effect that she regarded the possibility of using a basket in place of a cylinder in a USP type III apparatus as ridiculous. She was of the view that the reference to a basket in claim 1 was a mistake. When cross-examined in relation to the “Dissolution Basket Adapter for BIO-DIS” depicted in the VanKel documents she certainly did not suggest that it could not work or that the idea of making an adapter of that kind was ridiculous. Nor is her evidence inconsistent with the primary judge’s acceptance that the skilled addressee would not have dismissed the possibility of using a basket in place of a cylinder out of hand.
95 Of course, it does not follow that the skilled addressee, armed only with the patent specification and the common general knowledge as at the priority date, would be able to make a suitable adapter. But the primary judge did not make any finding to that effect. Rather, his Honour made two related findings: first, that it would be possible to use a basket in place of a cylinder in a USP type III apparatus, and second, that this possibility would not be dismissed by the skilled addressee at the priority date as ridiculous. In our view both of these findings were open.
96 GSK submitted that none of the expert witnesses were aware as at the priority date of any modification of the USP type III apparatus and that none were aware of the possibility of a basket being used in place of a cylinder in that apparatus. It was submitted that the primary judge’s construction of claim 1 therefore produces a result that, on the evidence of all the expert witnesses, could never have been in his or her contemplation at the priority date.
97 We think GSK’s submissions miss the point made by the primary judge. His Honour was not concerned with whether, as at the priority date, the skilled addressee was aware of any modification to the USP III apparatus or whether he or she may have previously contemplated making any such modification. His Honour’s point was that the skilled addressee, when confronted with the Patent and its reference to “the USP type III apparatus, reciprocating basket”, would not have dismissed as ridiculous the idea that this referred to a modified USP type III apparatus with a reciprocating basket.
Would a skilled addressee have known that a USP type 1 apparatus would readily fit into a USP type III vessel?
98 The primary judge found at [312] that a USP type I apparatus basket would readily fit into a USP type III apparatus vessel and that this would have been known to the skilled addressee. The various dimensions for the basket and the vessel are also set out in [312] of his Honour’s reasons. Since these dimensions are reproduced from the USP, which was common general knowledge at the priority date, the skilled addressee must be taken to have been aware of them. The inference that it would be open to the skilled addressee to perform the mental calculations necessary to perceive that a basket would readily fit into the vessel of a USP type I apparatus was one that the primary judge was entitled to draw.
Significance of the challenged findings
99 Although we have rejected GSK’s challenge to a number of factual findings, it should not be assumed that we regard them as material to the outcome of GSK’s appeal. We say this because the primary judge, despite his factual findings, considered that it was likely that the skilled addressee would have understood that the word “basket” was used in claim 1 in error. It follows that his Honour was not persuaded that his factual findings were determinative of the infringement case.
100 For reasons which we will now explain we think the primary judge was correct in holding that “basket means basket” despite the fact that the skilled addressee would understand that a mistake had been made in the drafting of the complete specification including, most importantly, the claims.
THE PRINCIPLES OF CONSTRUCTION
Background
101 There was no dispute between the parties as to the essential principles governing the construction of patent specifications. The differences between the parties concerned how those principles were to be applied in the circumstances of this case and in light of the primary judge’s findings as to how the skilled addressee would interpret the Patent.
102 The relevant principles have been summarised in many previous decisions of this Court: see, for example, Décor Corp Pty Ltd v Dart Industries Inc (1988) 13 IPR 385 (Sheppard J) at 400, Flexible Steel Lacing Company v Beltreco Ltd (2000) 49 IPR 331 (Hely J) at [70]-[81] and Jupiters Ltd v Neurizon Pty Ltd (2005) 222 ALR 155 (Hill, Finn and Gyles JJ) at [67]. Nevertheless, it is worth referring to some of these principles in more detail given that GSK’s principal submission is that they were misapplied by the primary judge.
103 Section 40(2) of the Act requires that the complete specification describe the invention fully including the best method known to the patentee of performing the invention, and end with a claim or claims that define the invention. The function of the claims is to define the scope of the patentee’s monopoly or, as it is sometimes described, the forbidden territory. The claims must be clear and succinct and fairly based on matter described in the specification.
104 A suitably qualified expert witness may give evidence on the meaning which persons skilled in the art would give to technical or scientific terms, and to any unusual or special meaning which they might give to words apart from their ordinary meaning. However, the construction of the specification and, in particular, the claims, is ultimately a matter for the Court to determine.
105 In Welsh Perrin Dixon CJ, Kitto and Windeyer JJ explained the function of the different parts of a complete specification and the rules that govern its proper construction. Their Honours observed at 610 that a patent specification is not to be read in the abstract but in light of the common general knowledge in the art before the priority date.
Purposive construction
106 More recent cases have continued to emphasise the need to read a patent specification as a whole and in light of the common general knowledge. They also confirm that a patent specification should be read in a practical and common sense way and given a “purposive” construction. This approach to construction requires the court to read the specification through the eyes of the skilled addressee with practical knowledge and experience in the field of work in which the invention was intended to be used and a proper understanding of the purpose of the invention.
107 A well-known case in which these principles were applied was the earlier decision of the House of Lords in Catnic Components Limited & Anor v Hill & Smith Limited [1982] RPC 183 (HL). The question in that case was whether the defendant had infringed the plaintiffs’ patent for a galvanized steel lintel that the claim required should have a rear support member “extending vertically” from the base plate. The defendant’s lintel was not precisely vertical, but instead extended upwardly at an angle of 84°. The House of Lords held that the defendant’s lintel was nevertheless within the claim. This was because the person skilled in the art would recognise that in order to perform the same function as the rear support member described in the claim, it was not essential that the rear support member in the defendant’s lintel be precisely vertical.
108 One criticism made of the decision of the House of Lords in Catnic was that it permitted the court to adopt an interpretation of a claim that travelled beyond what was conveyed by the language used so that the forbidden territory extended to devices with rear support members that were not truly vertical. However, as Lord Hoffmann later explained in Kirin-Amgen Inc v Hoechst Marion Roussel Ltd (2004) 64 IPR 444 at [34]:
“Purposive construction” does not mean that one is extending or going beyond the definition of the technical matter for which the patentee seeks protection in the claims. The question is always what the person skilled in the art would have understood the patentee to be using the language of the claim to mean. And for this purpose, the language he has chosen is usually of critical importance. The conventions of word meaning and syntax enable us to express our meanings with great accuracy and subtlety and the skilled man will ordinarily assume that the patentee has chosen his language accordingly. As a number of judges have pointed out, the specification is a unilateral document in words of the patentee’s own choosing. Furthermore, the words will usually have been chosen upon skilled advice. The specification is not a document inter rusticos for which broad allowances must be made. On the other hand, it must be recognised that the patentee is trying to describe something which, at any rate in his opinion, is new; which has not existed before and of which there may be no generally accepted definition. There will be occasions upon which it will be obvious to the skilled man that the patentee must in some respect have departed from conventional use of language or included in his description of the invention some element which he did not mean to be essential. But one would not expect that to happen very often.
109 It is important to note that Lord Hoffman was referring here to the meaning conveyed to the skilled addressee by the language used and was not directing himself to a situation in which the skilled addressee deduced that the language of the claim, although conveying to him or her a particular meaning, could never have been intended to mean what it conveyed.
110 Thus, a skilled addressee may understand a claim that required that something be “vertical” to mean “substantially vertical” or that a claim that includes a requirement that a device “prevents air from coming into contract with the surface of the ink” did not require that it prevent all the air from doing so: Henrikson v Tallon [1965] RPC 434 (HL). These are situations in which the court endeavours to give effect to the skilled addressee’s understanding of the claim language in preference to a purely literal or grammatical construction, not because the skilled addressee understands that the claim contains a mistake that requires correction, but because, when read in the context of the document as a whole and the common general knowledge, the words used would convey that meaning to the skilled addressee.
111 In the present case the primary judge found that the skilled addressee would understand that the reference to a reciprocating basket in claim 1 was an error and that he or she would have simply disregarded those words.
112 GSK relies on the skilled addressee’s understanding of the claim to establish not what the language of the claim actually conveys to the skilled addressee, but for the purpose of ignoring some of the language used on the basis that it must have been included by mistake. The question is whether, if the court were to also interpret the claim so as to correct the mistake, it would be re-writing the claim or merely interpreting it through the eyes of the skilled addressee in accordance with the principles to which we have referred.
Amendments
113 Before considering some additional authorities relied on by GSK, it is necessary to refer to some of the statutory provisions concerned with the amendment of patent specifications to correct errors.
114 Before 1883, the Master of the Rolls and the Lord Chancellor could allow amendments to patent specifications to correct clerical errors that were the result of mistake or inadvertence: Terrell on Patents, (5th ed, Sweet & Maxwell Limited, 1909) at p 172. Claims were until then optional, but became mandatory when the Patents Designs and Trade Marks Act 1883 (UK) (“the 1883 Act”) came into force.
115 The 1883 Act permitted a patentee to request that the Comptroller give leave to amend a patent specification “by way of disclaimer, correction or explanation”. It also required that the request be advertised, and permitted any person who gave notice of objection to be heard in opposition to the proposed amendments. No amendment was allowed if the specification, as amended, would claim an invention substantially larger than, or substantially different from, the invention claimed by the specification as it stood before amendment.
116 The 1883 Act also provided that the patentee could not recover damages for infringement occurring before an amendment was made unless the patentee established that “the original claim” was framed in good faith and with reasonable skill and knowledge.
117 The Patents Act 1990 (Cth), like its predecessors, contains similar, but somewhat more elaborate provisions for governing the amendment of patent specifications. The relevant provisions are found in Chapter 10 (ss 102-116) of the Act. Section 104 permits the Commissioner to allow amendments to a complete specification except those that are not allowable under s 102. Section 102 provides that an amendment is not allowable if, as a result of the amendment:
the specification would claim or disclose matter that extends beyond what is disclosed in the complete specification as filed (or certain other prescribed documents);
a claim of the specification would not in substance fall within the scope of the specification before amendment; or
the specification would not comply with s 40(2) or (3) of the Act.
Importantly, however, s 102 does not apply to an amendment for the purposes of correcting a clerical error or an obvious mistake made in, or in relation to, a complete specification.
118 Section 115 of the Act also restricts the right of the patentee to recover damages or profits in respect of any infringement of the patent before the date of any amendment. For the patentee to recover damages or profits in respect of any such infringement, the court must be satisfied that the specification without amendment was framed in good faith and with reasonable skill and knowledge.
119 Since GSK has not sought to amend the complete specification to correct what it contends is an error in the body of the specification and the claims, it is not appropriate for us to consider whether such an application would have been granted were it to have been made. An important question that would need to be determined is whether s 102 applied to the proposed amendments. That would involve a consideration of the scope of the claims in their current form and in the proposed amended form and, potentially, whether the error sought to be corrected was an “obvious error” or the result of a “clerical error”.
120 What is significant for present purposes is that the Act makes elaborate provision for the amendment of a complete specification. The relevant provisions limit the power to allow amendments in significant respects particularly where the proposed amendments have the effect of enlarging the scope of a claim. They also prevent a patentee who has amended its patent from obtaining any pecuniary relief for infringements occurring before amendment unless the specification before amendment was framed in good faith and with reasonable skill and knowledge.
121 We think these provisions of the Act are a powerful indication that a mistake in a claim cannot be ignored simply because the skilled addressee understands that a mistake has been made which he or she corrects by reading the language of the claim as if those words did not appear in it. The principles of construction must provide some basis which would permit the court to ignore the words of the claim. The mere fact that the skilled addressee would understand that they had been included by mistake is not sufficient.
The Authorities
122 GSK relied on a number of authorities concerned with the correction of errors in patent specifications which it contended supported its position.
123 The first of these was Simpson v Holliday (1865) 12 LT 99. That case concerned, amongst other issues, allegations that the patent was void for insufficiency and inutility because the specification described two different processes one of which required the application of heat and another that did not. In the Court of Appeal in Chancery, Lord Westbury LC, when addressing the defendant’s contention that the scope of the invention as described in the specification extended to a process that was useless, said at 99-100:
If a specification alleges a particular process, which may be slow, troublesome and expensive, is effective, and the statement is untrue, the vice is not removed by the fact that the same specification also describes another process which is efficient and which is stated to be speedy, certain and economical. When it is said that an error in a specification, which any workman of ordinary skill and experience would perceive and correct, will not vitiate a patent, it must be understood of errors which appear on the face of the specification, or the drawings it refers to; or which would be once discovered and corrected in following out the instructions given for any process or manufacture, and the reason is, because such errors cannot possibly mislead. But the proposition is not a correct statement of the law, if applied to errors which are discoverable only by experiment and further inquiry.
124 On appeal, Lord Chelmsford (with whom Lord Cranworth agreed) said at 320-321:
It was contended, on the part of the Appellants, that the word “or” in the specification ought to be read “and.” But there is nothing in the terms of the descriptions, nor upon the face of the specification itself, which would justify the Court in thus changing the form of the expression. It was also said that there was a considerable body of evidence to shew that skilled persons, to whom the specification must be taken to be addressed, found no difficulty in working it out, and applied heat in the process as a matter of course. This, however, cannot have any effect upon the construction of the specification. It merely proves that the description, though erroneous, is not likely to mislead skilled workmen. That the description may induce the necessity of experiments appears from the evidence of an experienced chemist, who says, “If I found there was no action without heat, I should heat it immediately.”
125 We do not think either of these statements of principles is of any real assistance in this case. Their Lordships were concerned with the sufficiency of the description of an invention and whether the patent might not be void if the skilled addressee could work out that it was necessary to perform the invention in a useful manner. Importantly, the fact that the skilled addressee might have no difficulty in working out that it was necessary to apply heat, and that the skilled addressee was not likely to be misled by the specification, was not considered sufficient to avoid the conclusion that the patent was void.
126 GSK also relied on the decision of Gummow J in Sartas No 1 Pty Ltd v Koukourou & Partners Pty Ltd (1994) 30 IPR 479, which is the decision that is cited by Mr Bodkin in support of the statement in his work to which we previously referred. In Sartas, claim 12 was to:
A spacer for a building foundation form work arrangement wherein a plurality of boxes are located in spaced apart relationship to define therebetween channels, the spacer including outermost engaging surfaces adapted to engage against both sides adjoining a corner of a first box, and at the same time against both corners [sides] adjoining a corner…
(emphasis added)
127 It was common ground before Gummow J that the word “corners” should read “sides”. His Honour said at 488:
One preliminary point should be mentioned. It concerns the fifty-first word in claim 12. This reads “corners”. If the claim is read in this form it is clear that there is no infringement. At the trial, in the end it was accepted by all counsel that “corners” should read “sides”. If the significance of the discrepancy concerned no more than the parties presently before the court, then there might be scope for application of a principle that no formal amendment was necessary because the true meaning was apparent as a matter of construction; cf Fitzgerald v Masters (1956) 95 CLR 420 at 426-7; Watson v Phipps (1985) 63 ALR 321 at 322; 60 ALJR 1 at 3.
But, of course, the specification operates to confer rights in rem and the legislation makes specific provision for amendment. Further, I was told that infringement litigation concerning the patent is on foot in another court against [third] parties …
Accordingly, on the last day of the trial, 2 September 1994, I gave directions for the advertisement of a motion seeking amendment of claim 12. The notice of motion will be made returnable no earlier than 14 days after delivery of these reasons for judgment. In the meantime, it was accepted that I should proceed to determine this case on the footing that claim 12 uses the term “sides” rather than “corners”.
128 His Honour, who delivered reasons for judgment on 2 December 1994, found that claim 12 was not infringed and was also invalid for reasons that had nothing to do with the error in that claim. However, he indicated at 512 that he would not make final orders until after he had disposed of the motion seeking the amendment of claim 12.
129 An appeal to the Full Court was allowed in part but not in relation to the infringement issue: Leonardis v Sartas No 1 Pty Ltd (1996) 67 FCR 126 (Burchett, Hill and Tamberlin JJ). The Full Court described the error in claim 12 as “an obvious falsa demonstratio” and noted that Gummow J made an order on 19 December 1994 amending the patent to correct the error. Our review of the Court’s records confirms that his Honour made orders on 19 December 1994 correcting the error in claim 12 before making an order revoking the patent.
130 We do not think that Sartas assists GSK’s position. On the contrary, the procedure followed by Gummow J in that case is consistent with the view that the error in claim 12 could only be corrected by amendment.
131 The next decision relied on by GSK was that of Judge Birss QC (as he then was) in Lizzanno Partitions (UK) Limited v Interiors Manufacturing Limited [2013] EWPCC 12. The case concerned a patent for internal glass partitions that included a gasket comprising hollow tubing made from a “non-rigid” material. His Honour noted that the specification stated that the hollow tube could be made of any suitable non-rigid material and that usually the hollow tube will comprise a plastic material such as uPVC (an abbreviation for unplasticised PVC). But his Honour also found that this would be potentially confusing as no skilled addressee would regard uPVC as a non-rigid material. Indeed, his Honour found that “[n]o-one would think that the patent really meant that one could use uPVC.”
132 Claim 10 required that the gasket be made of uPVC. On the question of the infringement of this claim, Judge Birss QC said at [78]:
The argument is I think that because it is obvious that the references to uPVC in the specification are a mistake, the true interpretation of claim 10 is that it is a claim to PVC and not uPVC. I do not propose to spend much time on this. I do not agree. It is one thing to read the description fairly, realise that the reference to uPVC there must be the result of an error and so read the document as a whole bearing in mind that uPVC is not flexible plastic; it is quite another to come to a conclusion that a patent claim to uPVC actually means something else. For that to get off the ground the correction of the mistake would have to be clear as well but it is not. Perhaps the patentee genuinely thought uPVC was flexible and meant to claim it. This would still not affect the overall interpretation of the patent but it would not justify reading claim 10 as if it was a claim to PVC. I reject the allegation of infringement of claim 10.
133 Properly understood, this case does not assist GSK. Judge Birss QC was unwilling to read uPVC to mean PVC in claim 10 even though no skilled addressee would understand the patent to teach the use of uPVC in the invention described in the specification.
134 GSK also relied on a decision of the High Court of Eire in Farbwerke Hoechst Aktiengesellschaft v Intercontinental Pharmaceutical (EIRE) Limited [1968] FSR 187. In that case Kenny J concluded that there was an error in dependent claim 8 which referred to a “formula R1 – NH2 in which R and R1 have the meanings given above”. In fact, claim 1 (on which claim 8 was dependent) defined R1, R2 and R3 but not R. Kenny J said at 189:
As there is no R in any of the claims, the reference to it in claim 8 is an obvious error and the sentence should read “in which R1, R2 and R3 have the meanings given above”. This error did not mislead any of the expert scientists who gave evidence or who have acted as counsel in this case and does not invalidate the patent (see the speech of Lord Westbury in Simpson v Holliday (1866) L.R. 1 H.L. 315) [sic].
135 We do not doubt that the result in Farbwerke Hoechst was correct, but not because the error would not mislead the skilled addressee. It seems to us to be a perfectly reasonable construction of claim 8 to say that R1, R2 and R3 are to take their meaning from claim 1. While it is true that R was not defined in claim 1 (or anywhere else it seems), it is also true that R was not itself used in the formula in claim 8. So the definition of R in claim 8 was truly otiose, and could be disregarded on that basis. We do not think that approach involves any re-writing of the claims. The position would be quite different if R had been used in the formula.
HOW SHOULD CLAIM 1 BE CONSTRUED?
136 As the primary judge held, “basket” means “basket” not “cylinder”. If claim 1 was intended to refer to the compendial apparatus then it would not be necessary to say more than “the USP III apparatus”. The elaboration that follows these words in the claim strongly suggests that a simple reference to “the USP type III apparatus” was not sufficient to describe the relevant apparatus. On GSK’s construction of the claim, not only is the word “basket” an error, but the word “reciprocating” is otiose.
137 Further, it is clear that the words “the USP type III apparatus, reciprocating basket” are not used to describe anything new if what is being referred to is the compendial USP type III apparatus. So this is not a case in which it can be argued that the problem with the language used has anything to do with the difficulties that may face a patent applicant attempting to describe something new.
138 GSK’s infringement case can only succeed if the words “reciprocating basket” are either interpreted to mean “reciprocating cylinder” or simply ignored. Either approach involves an impermissible re-writing of the relevant claims.
139 This is a case in which the skilled addressee’s understanding of the claims, as found by the primary judge, does not make proper allowance for the function of the claims in defining the invention. Ultimately, it is for the court to decide the meaning of the claims. This is a case in which we think the language of the claim must be understood to mean what it actually says.
140 On its proper construction claim 1 refers to the USP III apparatus with a reciprocating basket.
141 The respondents contended that if claim 1 was to be understood as referring to the USP III apparatus with reciprocating cylinder (ie. the compendial device) then the relevant claims were invalid for lack of clarity. However, they made it clear that they only relied upon this contention in the alternative. It is not necessary for us to consider it given our interpretation of the relevant claims.
142 In our view GSK’s appeals should be dismissed.
THE CROSS APPEALS
143 The cross appeals challenge the primary judge’s rejection of the respondents’ cross-claims seeking orders for revocation of the patents on the grounds that the relevant claims were not fairly based on matter described in the complete specification, that there had been a failure to disclose the best method known to the patent applicant of performing the invention, and that the relevant claims were not clear and succinct and failed to define an invention. The latter ground was said to be relied on only if we were of the view that the primary judge had misconstrued the claims.
144 Section 40(2) and (3) of the Act (in the form it took immediately prior to its amendment by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012) relevantly provides:
(2) A complete specification must:
(a) describe the invention fully, including the best method known to the applicant of performing the invention; and
(b) where it relates to an application for a standard patent—end with a claim or claims defining the invention; and
…
(3) The claim or claims must be clear and succinct and fairly based on the matter described in the specification.
Fair Basis
Background
145 The primary judge summarised the relevant principles at [600]-[603] before recording that they were not in dispute. In the cross-appeal, Apotex accepted the primary judge’s summary of the relevant principles as accurate, but contended that they were misapplied by his Honour.
146 Apotex submitted that the primary judge did not undertake the required textual analysis of the specification to determine whether the claims travel beyond the matter described in the specification and, in addition, that his Honour made the following related errors:
he relied on expert evidence and documents outside the specification to supplement the disclosure of the invention;
he approached the question of fair basis, in effect, by asking whether the breadth of the claims was justified or reasonable; and
he relied on matters going to utility and sufficiency.
147 Apotex submitted the relevant claims did not comply with s 40(3) of the Act because the broad range of the claimed pharmaceutical compositions “travels beyond” the matter described in the specification. It submitted that the body of the specification read as a whole discloses as the invention only two particular formulations (Formulations C and D) which the primary judge in any event treated as being essentially the same. Thus, the invention described in the specification, according to the submission, is essentially a single pharmaceutical composition of paracetamol in a two-phase formulation that has a “unique” in vitro dissolution profile.
148 As to the consistory clauses which mirror the language of (inter alia) claim 1, Apotex submitted that these were contrary to the description of the specification when read as a whole and could not of themselves provide fair basis for any of the relevant claims.
149 It is common ground that the relevant claims cover a much broader range of pharmaceutical compositions than Formulations C and D. This is apparent from the following table produced by Apotex:
The relevant principles
150 Section 40(3) of the Act requires that a claim be fairly based on the matter described in the specification. The “invention” in this context refers to “the embodiment which is described and around which the claims are drawn”: Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 (“Lockwood”) at [53], [54], [69] and [77] approving AMP Inc v Utilux Pty Ltd (1971) 45 ALJR 123 at 127 per McTiernan J.
151 When applying s 40(3) of the Act, the question is:
… whether the claim is fairly based is not to be resolved … by considering whether a monopoly in the product would be an undue reward for the disclosure. Rather, the question is a narrow one, namely whether the claim to the product ... as expressed, travels beyond the matter disclosed in the specification.
per Barwick CJ in Olin Corporation v Super Cartridge Co Pty Ltd (1977) 180 CLR 236 at 240 referring to s 40(3) of the Patents Act 1952 (Cth) (“the 1952 Act”) approved in Kimberly Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [15] as re-affirmed by the High Court in Lockwood at [57]. In Lockwood, the High Court also said in reference to s 40(3) of the 1952 Act at [68]-[69]:
[68] … The comparison which s 40(3) calls for is not analogous to that between a claim and an alleged anticipation or infringement. It is wrong to employ ‘‘an over meticulous verbal analysis’’. It is wrong to seek to isolate in the body of the specification ‘‘essential integers’’ or ‘‘essential features’’ of an alleged invention and to ask whether they correspond with the essential integers of the claim in question.
[69] ‘‘Real and reasonably clear disclosure’’. Section 40(3) requires, in Fullagar J’s words, ‘‘a real and reasonably clear disclosure’’ [… in Société des Usines Chimiques Rhône-Poulenc v Commissioner of Patents (1958) 100 CLR 5 at 11 … ]. But those words, when used in connection with s 40(3), do not limit disclosures to preferred embodiments.
‘‘The circumstance that something is a requirement for the best method of performing an invention does not make it necessarily a requirement for all claims; likewise, the circumstance that material is part of the description of the invention does not mean that it must be included as an integer of each claim. Rather, the question is whether there is a real and reasonably clear disclosure in the body of the specification of what is then claimed, so that the alleged invention as claimed is broadly, that is to say in a general sense, described in the body of the specification.’’
Fullagar J’s phrase serves the function of compelling attention to the construction of the specification as a whole, putting aside particular parts which, although in isolation they might appear to point against the ‘‘real’’ disclosure, are in truth only loose or stray remarks.
(some citations omitted)
152 A consistory clause may provide a fair basis for a claim which uses the same language but not if there are other matters disclosed in the specification which show that the invention is different from what the consistory clause suggests. The High Court also said in Doric at [87] and [99]:
[87] Finally, it is necessary to consider the trial judge’s citation of Atlantis Corporation Pty Ltd v Schindler [(1997) 39 IPR 29] for the proposition that to couch a claim “in the same terms as the description of the invention in the specification” did not of itself, by that mere ‘‘coincidence of language’’, establish fair basing. That proposition is correct, but it is not fatal to the Patentee’s position in this case. A “coincidence of language” between a claim and part of the body of a specification does not establish fair basing if that part of the language of the specification does not reflect the description of the invention in the light of the specification as a whole ...
[99] ... the correct position is that a claim based on what has been cast in the form of a consistory clause is not fairly based if other parts of the matter in the specification show that the invention is narrower than that consistory clause. The inquiry is into what the body of the specification read as a whole discloses as the invention [Welch Perrin & Co Pty Ltd v Worrel (1961) 106 CLR 588 at 612–613]. An assertion by the inventor in a consistory clause of that of which the invention consists does not compel the conclusion by the court that the claims are fairly based nor is the assertion determinative of the identity of the invention. The consistory clause is to be considered by the court with the rest of the specification.
153 These principles were applied by Bennett, Nicholas and Yates JJ in Sigma Pharmaceuticals (Australia) Pty Ltd v Wyeth (2011) 119 IPR 194 which concerned claims for extended release formulations of venlafaxine hydrochloride and methods of treatment that employed these formulations. It was held that the claims covered formulations of venlafaxine hydrochloride made using a particular type of tableting technology even though the specification, when read as a whole, indicated it was not suitable for use in making the formulations of the invention. It was in those circumstances that the Full Court held that the broad consistory clauses in the specification did not provide a fair basis for the claims.
Consideration
154 It is important to note that it was Apotex’s submission to the primary judge that the consistory clauses could not provide the required fair basis for the claims because the skilled addressee would understand that the invention described was in fact narrower than the consistory clauses suggested. All that the specification when read as a whole disclosed, according to this submission, was a single formulation (or two formulations that were essentially the same) with a defined dissolution profile. To succeed in this case Apotex needed to show that there is other matter included in the specification which shows that the invention that is described in the specification as a whole is narrower than what is described in the consistory clauses.
155 In support of its submission Apotex placed great weight on the statement appearing at page 4 lines 11 to 14 (referring to the formulation with “a unique in vitro dissolution profile”) as indicating that the specification was directed to a single formulation with a specific dissolution profile.
156 We do not understand the description of the invention to be so limited. The passage at page 4 lines 11 to 14 must be read in the context of the specification as a whole (including the consistory clauses) which suggests that, contrary to Apotex’s submission, the invention described is not limited to the exemplified embodiments, Formulations C and D. Those formulations are merely preferred embodiments which are used, as stated at page 8 lines 16 to 17 of the Patent, to “illustrate the advantageous properties of the compositions of the present invention”. These preferred formulations are also described at page 12 lines 5 to 6 and page 15 lines 12 to 13 as “having an in vitro dissolution profile falling within the scope of the present invention.” None of these statements is consistent with the description of an invention the scope of which is limited to a single formulation.
157 In our view the invention described in the Patent is a bi-layer tablet having a ratio of immediate release (“IR”) phase of paracetamol and sustained release (“SR”) phase of paracetamol, an in vitro dissolution profile within the ranges summarised on pages 4a and 5 of the Patent, and a matrix forming polymer in the SR phase.
158 There was evidence before the primary judge which indicated that formulations with a paracetamol content within ±10% of the ideal value or label claim, and a dissolution profile within ±10 percentage points of the preferred dissolution profile, were likely to be bioequivalent to the preferred formulation. His Honour found at [651] that changes within those ranges were unlikely to have a significant clinical effect. That finding was not challenged by either of the respondents.
159 The primary judge referred to documents published by the FDA relevant to the skilled addressee’s understanding of the relationship between the dissolution profiles specified in the consistory clauses for the purpose of ascertaining what the specification, when read as a whole, disclosed to the skilled addressee. These documents included an FDA report that gives guidance for dissolution ranges for the purpose of quality control between batches of the same formulation. It was not disputed that these documents formed part of the common general knowledge at the priority date.
160 Apotex criticised the primary judge for taking into account material that is not referred to in the Patent. Apotex relied on the following statement in the High Court judgment in Lockwood at [48]:
If all that is essential in assessing a fair basing objection is recourse to the contents of the specification, there is no call, for example, for an examination (except on construction questions) of common general knowledge (which is essential when considering an objection based on want of an inventive step), or of prior art (which is essential when considering novelty (s 7(1))) …
161 This statement was relied on by Apotex in support of the proposition that it is impermissible to rely on common general knowledge when determining whether a claim is invalid for lack of fair basis. However, the High Court was not suggesting that the common general knowledge cannot be relevant to the issue of fair basis. It recognised that the issue of fair basis is often bound up with issues relating to the proper construction of the specification. Thus, it may be necessary to have regard to the common general knowledge for the purpose of construing the specification and, in particular, characterising the invention described when determining whether the invention claimed is fairly based on the matter described in the specification.
162 In the present case, the FDA documents were directly relevant to the question whether the Patent, when read in light of the common general knowledge, describes an invention comprising a single formulation even though, as found by the primary judge, the skilled addressee would understand that the pharmacokinetic behaviour of at least some of the other formulations with paracetamol content and a dissolution profile within the scope of the ranges specified in the consistory clauses and claims were likely to be bioequivalent and unlikely to differ significantly in clinical effect.
163 Apotex also criticised some of the primary judge’s language including at [651] where his Honour said that the FDA guidelines provide “a reasonable basis” for the claims. We do not think these criticisms have any substance. These remarks by his Honour must be understood in the context of Apotex’s submission that there was no basis for inferring that other formulations within the claims would have a pharmacokinetic profile resembling that of Formulations C and D unless there was an established in-vitro in-vivo correlation (“IVIVC”) that would enable one to safely predict the pharmacokinetic behaviour of one formulation based on that of another formulation. We do not think that his Honour’s remarks were inapposite given the submission to which they were responsive.
164 For a similar reason Apotex’s criticism of the reference by the primary judge at [653] to the promise of the invention as confusing the test for fair basis with that of inutility must be rejected. There his Honour was addressing the submissions that the teaching of the specification did not involve a reasonable extrapolation from Formulations C and D. It is apparent from the reasoning at [654]–[658], and made particularly clear by his observation at [658] that Apotex’s case amounted to speculation that the claims are likely to include formulations that do not possess the attributes identified in the Patent, which considerations might be relevant to utility, but are not to fair basis. Plainly enough the question of whether or not the claims meet the promise of the invention does not form part of the test for fair basis.
165 Apotex also submitted that the primary judge misunderstood the FDA guidelines because they give guidance for dissolution ranges for the purpose of quality control between batches of the same formulation. It was submitted that the FDA guidelines do not permit deviations of ±10% around the dissolution profile of a range of formulations beyond a single tested formulation.
166 What is critical to the pharmacokinetic behaviour of the many formulations within the claims is the dissolution profile (or release rate) of the formulation. The primary judge accepted that the FDA Report recognised that a variation of ±10% percentage points in the release rate was acceptable to the FDA even where no IVIVC had been established. This provides evidentiary support for the finding that the skilled addressee would know that various formulations within the claims apart from Formulations C and D were likely to have similar pharmacokinetic properties. This also provides a complete answer to Apotex’s argument that the skilled addressee (equipped with the common general knowledge) would approach the Patent with an understanding that there would be no reason to think that other formulations within the claims would have a similar pharmacokinetic profile to Formulations C and D in the absence of any established IVIVC.
167 Apotex also submitted that even if the disclosure in the specification is not limited to Formulations C and D, the ranges are not fairly based in the absence of an explanation of the mechanism or reasoning founding the relationship between those ranges and the pharmacokinetic parameters. It argued that for the claims to be fairly based, the specification must disclose an established IVIVC to provide a “predictive connection”. We do not accept this submission.
168 The specification does not need to identify the reasoning or the “theoretical basis” underpinning the selection of content, ratio and dissolution limits referred to in the claims. Similarly, for the purpose of providing fair basis, the specification does not need to describe the invention in a different or more detailed way than the claims. In Photocure ASA v Queen’s University at Kingston & Anor (2005) 216 ALR 41 Merkel J responded to a like submission at [136]:
In effect, the submission made by PhotoCure was that the examples provide the only “real and reasonably clear disclosure” in the specification. The difficulty with that qualitative approach is that in Lockwood at [87] the High Court observed that it is possible for a claim to be fairly based on nothing more than a use of the same words in the specification (including in a consistory clause), as long as those words reflect the disclosure provided in the specification when read as a whole. Thus, s 40(3) does not require that the specification describe the invention in a different or more detailed way than the supported claims.
169 We agree with Merkel J’s observations concerning s 40(3) of the Act as it stood prior to its amendment by the Intellectual Property Laws Amendment (Raising the Bar) Act 2012. We do not express any view in relation to s 40(3) of the Act in its current form.
170 Of course, it is important to note that s 40(2)(a) requires that the complete specification “describe the invention fully”. A complete specification may still “describe the invention fully” without explaining why the invention works. After all, the inventor, who presumably believes that the invention described works, may not understand why it works. But this does not prevent him or her from obtaining patent protection for the invention.
171 In the result, we agree with the primary judge that a skilled addressee would not understand that the Patent (contrary to what appears in the consistory clauses) discloses an invention consisting of a single formulation. Nor do we think there is anything disclosed in the specification as a whole to indicate that the invention described is narrower than what was described in the consistory clauses. In our view the appeal against the primary judge’s rejection of this aspect of Apotex’s case fails.
Best Method
Background
172 The respondents contended that the primary judge erred by failing to find that relevant claims were invalid on the ground that, at the time it was filed, the complete specification did not disclose the best method of performing the invention known to the patent applicant. In particular, they contended that the primary judge wrongly held that the best method requirement imposed by s 40(2)(a) of the Act could be satisfied if the skilled addressee could arrive at the best method known to the patent applicant by performing “routine experimentation” that did not require “ingenuity or undue experimentation.” The respondents contended that the complete specification must not leave the skilled addressee to conduct any experimentation that it may be necessary to perform in order to arrive at the best method known to the patent applicant.
173 It is necessary to begin our consideration of this ground of the cross-appeal by referring to a number of the primary judge’s findings.
174 The primary judge accepted at [749] that the patent applicant used a particular grade and viscosity of HPMC and a particular granulation end point to make Formulations C and D; that Formulation D had an in vitro dissolution profile that very closely resembled what the patent applicant considered to be the ideal release profile intended to deliver the ideal in vivo pharmacokinetic properties; that the patent applicant had tested Formulations C and D in humans and found that those formulations worked; and that the method of making Formulations C and D disclosed in the complete specification described a method that used high viscosity HPMC and a conventional method of granulation.
175 The primary judge accepted at [838] that the patent applicant was aware that the selection of the grade and viscosity of the high viscosity HPMC and granulation endpoint used to make Formulations C and D would affect the in vitro dissolution profile of the tablets by “moving away from the ideal release profile”. His Honour also accepted at [839] that the patent applicant knew that some high viscosity HPMCs and granulation endpoints were not as good as others at least in the sense of achieving an in vitro dissolution profile that had little variation within and between batches and most closely aligned with the “ideal” in vitro profile.
176 The primary judge also accepted at [840] that the dissolution profile of the formulation affected its pharmacokinetic performance and that changes in the in vitro dissolution profile would affect its pharmacokinetic performance. However, his Honour also accepted Professor Davies’ evidence that a change in the in vitro dissolution profile of less than 10% would not result in a significant difference in the pharmacokinetic attributes. In those circumstances, the plasma profile would be “bioequivalent”.
177 His Honour said at [751]:
[751] In my view, the patent applicant disclosed the best method known to it of performing the invention by disclosing Formulations C and D in sufficient detail to enable a skilled addressee to make the same. Formulations C and D were themselves the best method to make or perform the invention known to the applicant. Not only that, but the experts in this case in fact agreed that they could make Formulations C and D from what had been disclosed in the specification. The respondents’ attack can be reduced to little more than saying that there has been a failure to disclose the best method of making the best method of performing the invention. That compounded idea and ideal is not the statutory requirement for disclosure.
(original emphasis)
178 Pausing there, we do not fully understand the last two sentences in [751]. In any event, it is apparent his Honour found that Formulations C and D as specified in the table appearing at page 12 and the additional description at page 15 lines 20 to 22 of the complete specification, disclosed to the skilled addressee the best method to perform the invention known to the patent applicant.
179 The primary judge also said at [842]-[844]:
[842] A patent will satisfy the best method requirement if the skilled addressee can arrive at the best method known to the patent applicant of performing the invention by some “routine” experimentation, but without ingenuity or undue experimentation. A patent applicant does not need to disclose details of the method which would be well-known and understood by the skilled addressee such as well-known analytical agents, commonly used methods, well-known terms of art, or a description of machinery in standard use. But a complete specification must disclose each essential element or feature for performing the invention, even if a skilled addressee would know or could readily ascertain that element.
[843] Where the best performance of the invention requires a step that is omitted by the complete specification, even if it could be readily ascertained by a skilled addressee, that will not meet the best method requirement.
[844] If the skilled addressee is left to make a choice as to what analytical agent to use, what commonly used method to use or what machinery to use or is left in doubt as to what a term means, the best method will not have been described if that choice or uncertainty affects the performance of that method.
180 His Honour accepted evidence given by Professor Davies that selecting a grade of HPMC to make Formulation C was a matter of routine. His Honour said at [849]:
[849] Further, Professor Davies explained that he would start with “K4M” HPMC, which had a viscosity of between about 3000 to 5000 cps, if he was making Formulation C, given that he was aiming for a release profile over about three hours in vitro. Professor Davies had a “good feeling” that K4M HPMC would work to produce Formulation C. He noted that he had used higher viscosity HPMC grades in formulations which release over much longer time periods. Professor Davies explained that if K4M HPMC did not work, he would move to the next higher-viscosity grade, but “there are only a limited number of grades of these that will effectively work”. I accept Professor Davies’ evidence that selecting a grade of HPMC to make Formulation C was a matter of routine.
181 It is clear that what is envisaged by this finding is that the skilled addressee would prepare a Formulation C using K4M HPMC or some other high viscosity HPMC as his or her starting point before, if necessary, preparing Formulation C using a different grade of HPMC. The skilled addressee then would work through the different grades of HPMC until he or she produced the dissolution profile and, therefore, the pharmacokinetic attributes, exhibited by Formulation C. The process involved is one involving classic trial and error, moving from one grade of high viscosity HPMC to the next until the desired grade is identified.
182 At [860]-[862] of his reasons the primary judge said:
[860] … GSK was not required to set out in the specification of the Patent manufacturing optimisation details developed with respect to its own particular equipment and manufacturing capabilities. In my view, such idiosyncratic detail is not relevant to the skilled person’s task of performing the invention claimed and is unlikely to assist a skilled person to perform the invention using his own particular excipients, manufacturing methods and equipment.
[861] Furthermore, I am unable to conclude on the evidence that formulations made with grades of HPMC optimised for the patentee’s own equipment are any “better” than other formulations which a skilled person would make when reproducing Formulation C/D.
[862] Finally … Professors Davies and Tucker considered that they could make Formulations C or D without undue experimentation. As discussed above, the Patent provided sufficient information for the skilled formulator to make formulations which acceptably replicated the dissolution profiles of both Formulations C and D. Accordingly the best method known by the patent applicant was disclosed in the Patent.
The relevant principles
183 Section 40(2)(a) of the Act (in its relevant form) imposes two distinct but closely related requirements with respect to the complete application. First, the complete specification must describe the invention fully. The test for determining whether this particular requirement (the sufficiency requirement) has been complied with as explained by the High Court in Kimberly Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [25] is encapsulated by the following question:
The question is, will the disclosure enable the addressee of the specification to produce something within each claim without new inventions or additions or prolonged study of matters presenting initial difficulty?
(footnote omitted)
See also Lockwood Security Products Pty Ltd v Doric Products Pty Ltd (2004) 217 CLR 274 at [60] and Tramanco Pty Ltd v BPW Transpec Pty Ltd (2014) 105 IPR 18 at [205]-[206].
184 The second requirement imposed by s 40(2)(a) (the best method requirement) is that the complete specification describe the best method known to the patent applicant of performing the invention at the date of filing.
185 The best method requirement has a long history in patent law which predates the introduction in 1932 of s 25(2)(j) into the Patents and Designs Act (1907) (UK) which made it a distinct statutory ground of revocation. As Fletcher Moulton LJ explained in Vidal Dyes Syndicate Ltd v Levenstein Ld. [1912] 29 RPC 245 at 269:
It is settled law that a patentee must act towards the public uberrimȃ fide, and must give the best information in his power as to how to carry out the invention. He is therefore bound to tell the public all the steps that can advantageously be taken in carrying out the invention.
186 The patent applicant is required to disclose information not already known to the skilled addressee by way of the common general knowledge that is required to perform the invention in the best manner known to the patent applicant as at the date the complete specification is filed.
187 Whether there has been a failure to make the required disclosure is essentially a question of fact. Every case will depend on its own facts including the nature of the invention, and the significance of what is and what is not disclosed. And like many other questions that arise in relation to the interpretation of a patent specification and the scope of its disclosure, the question whether there has been sufficient disclosure of the best method should be addressed in a practical and common sense manner. It is also necessary to have regard to the public policy justification that supports the best method requirement.
188 The primary judge referred in detail to the decision in Les Laboratoires Servier v Apotex Pty Ltd (2016) 247 FCR 61. In that case the invention claimed was the arginine salt of perindopril. The complete specification identified L-arginine salt as the most preferred L-arginine salt. It also stated that L-arginine salt was prepared according to “a classical method of salification of organic chemistry” but did not provide any other detail as to which method could be employed even though this was known to the inventor at the time it was filed. Nor did the specification disclose that the preferred L-arginine salt described was a crystalline salt, something that was also known to the inventor at the time the specification was filed. The trial judge found that the mere reference to a method of classical salification was inadequate because this left open too many variables to allow the skilled addressee to follow a routine process of deduction leading to the preferred salt.
189 The Full Court said at [134]-[137]:
[134] Perindopril arginine is generally a more stable product than perindopril erbumine [another perindopril salt that was prior art] and the claim is not to a specific form of the arginine salt. If Servier knew of a method that provides a form of the salt with the characteristics exemplified in the Patent, which characteristics provided the stated advantages of the invention over the prior art, it was incumbent on it to provide that method. This would relieve the skilled worker from making the choices within those necessarily made or available in a classical salification. The disclosure of the method known to Servier would not only have relieved the skilled addressee of confronting blind alleys and pitfalls which may not be uncommon in a general sense but also, and importantly, would tell the skilled addressee the methodology to achieve the form that obtains the result which constitutes the invention, that is increased stability and storage length. While claim 1 does not refer to any particular form of perindopril arginine, crystalline or otherwise, if Servier had a method that produced a product that was at least sufficiently crystalline or in a sufficiently good form so that it could be used in the API for the tablets used in the stability study described in the specification, that is precisely what should have been disclosed.
[135] Accepting that there was no lack of sufficiency, the mere fact that a complete specification described a method which conveyed sufficient information to a skilled addressee to enable him or her to work the invention does not necessarily satisfy the Patentee’s additional obligation to describe the best method. The patentee has an obligation to include aspects of the method of manufacture that are material to the advantages it is claimed the invention brings.
[136] In the present circumstances, the inventor, Mr Damien, had not characterised the products of the two methods that he utilised but he did know that those methods created the arginine salt in a useable form and, as a person skilled in the art, he knew that there were many alternatives available from which to choose. As the skilled worker, he knew that the method of classical salification was sensitive to choices such as the choice of solvent. He knew that some were likely not to be as good as others. That is consistent with the expert evidence, although it was not specifically put to Mr Damien.
[137] The method of making the perindopril arginine affects the form of that compound and that of the properties of the compound itself, including its stability and usability of formulation. Its making can also involve unnecessary choices and difficulties.
The Full Court accepted that the complete specification did not provide the skilled addressee with sufficient information to enable him or her to produce the preferred salt in crystalline form or at least not without engaging in what would amount to undue experimentation. Nor did the complete specification tell the skilled addressee that the full benefit of the invention could only be obtained by producing the preferred salt in crystalline form.
Consideration
190 The respondents submitted that the first sentence of [842] in the primary judge’s reasons was wrong and did not represent the law. In particular, it was submitted that his Honour wrongly conflated the test for determining compliance with the sufficiency requirement and the test for determining compliance with the best method requirement. We do not think this criticism is justified, especially when read in the context of [842] as a whole. The reference to “undue experimentation” suggests to us that his Honour accepted that there may be some situations in which the patent applicant will be justified in expecting the skilled addressee to perform some experimentation and some situations in which it will not. What constitutes “undue experimentation” in the context of the best method requirement will depend on the circumstances, including the qualifications, knowledge and capabilities attributed to the notional skilled addressee.
191 We do not think the patent applicant is entitled to withhold information that is necessary to enable the skilled addressee to perform the invention in accordance with the best method merely because the skilled addressee could ascertain such information by routine experiment. There is a great deal of experimentation performed in the field of drug development and formulation which, although routine and not requiring the application of any ingenuity, may be, time consuming and expensive. For the patent applicant to withhold information which it knows is necessary to perform the invention in accordance with the best method merely because the information could be obtained by routine experimentation is in our view inconsistent with what Fletcher Moulton LJ described as the settled law that required the patentee to give the best information in its power as to how to carry out the invention.
192 Whether or not it will be open to the patent applicant to not disclose relevant information on the basis that it is available to the skilled addressee by routine experimentation will depend on the importance of the information in question, the practicality of disclosing it, and the extent of the burden imposed on the skilled addressee who is left to rely upon routine experimentation. That question is, as we have already mentioned, to be addressed in a practical and common sense manner.
193 The crucial question that arises in this part of the appeal is whether the complete specification satisfies the best method requirement even though it does not disclose the particular grade or viscosity of the high viscosity HPMC or the granulation end points used by the patent applicant to make Formulations C and D.
194 The relevant claims include in their definition of the sustained release phase of the claimed tablet a matrix forming polymer. The purpose of the matrix forming polymer is to provide the tablet with a particular dissolution profile within the range claimed which for Formulation C is as shown in Table 2 at page 13, and which for Formulation D is as shown in Table 3 at page 15. The matrix forming polymer is a crucial component of the formulations claimed which plays an essential role in achieving the desired dissolution profile.
195 Formulations C and D are the embodiments of the invention described in the complete specification and around which the claims are drawn: Kimberly Clark Australia Pty Ltd v Arico Trading International Pty Ltd (2001) 207 CLR 1 at [19] citing AMP Inc v Utilux Pty Ltd (1971) 45 ALJR 133 at 127 per McTiernan J. It is by fully describing such embodiments that a patent applicant will usually seek to comply with the best method requirement. This is the approach taken by the patent applicant in this case. Formulations C and D, which each have their own unique dissolution profile, are presented by the patent applicant as the most preferred embodiments of the invention and are described in the most detail.
196 The respondents based their case on documents of the patent applicant which showed the grade and viscosity of the “high viscosity HPMC” used to prepared Formulations C and D (HPMC 2208 4000 cP). These documents included a marketing authorisation application (“the MAA”) dated May 2000, approximately 12 months prior to the date upon which the complete specification was filed.
197 The MAA includes Section 4.1 headed “Product Development” which begins with a discussion of the need for extended release paracetamol that may improve a patent’s quality of life by reducing the number of doses taken and by providing steadier levels of drug in plasma and improved control of pain. The MAA continues:
The objective of this product development was to develop a paracetamol tablet that could provide a rapid analgesic effect that would be sustained for up to 6 to 8 hours. To achieve this objective it was considered that the product should provide a pharmacokinetic profile that should meet the following criteria:
1. Therapeutically active drug plasma concentrations should be attained rapidly.
2. The maximum plasma concentration (Cmax) should not exceed those of normal adult oral dose formulations (two 500 mg tablets).
3. The average plasma concentration at 6 h following a single dose should not be less than the minimum therapeutic level of 4 mcg/ml (Hossain 1992) (3).
4. The extent of absorption should be equivalent to conventional IR paracetamol tablets i.e. all of the dose should be released within the residence time of the tablets in the GI tract which would be limited to 12 hours post dose. No appreciable absorption would take place beyond this time point.
A bi-layer tablet design was considered suitable to achieve these objectives. Using this approach it was possible to produce a product that demonstrated an initial release of paracetamol from the IR layer followed by slow release of paracetamol from the matrix SR layer which contained a high viscosity grade of HPMC (2208 4000 cP) to control the rate of diffusion of paracetamol from the dosage form. The release profile from such a dosage form could then be modified by either altering the IR to SR ratio or by altering the amount of HPMC in the SR formulation.
198 More information concerning the grade and viscosity of the HPMC used appears in Section 4.2.2 of the MAA. Under the heading “Sustained Release Layer” the MAA states:
Hydroxypropyl Methylcellulose 2208 4000 cP (HMPC) is a water soluble polymer, which is frequently used in the formulation of controlled release dosage forms (Handbook of Pharmaceutical Excipients 1986)(10). The mechanisms by which it retards drug release include its ability to rapidly hydrate and form a gel layer at the matrix periphery; the drug is then released from the matrix by a combination of diffusion through, and erosion of, the gel layer. The type of HPMC used as well as the water solubility of the draft, will influence the rate of diffusion and erosion.
Among different grades of HPMC, which are commercially available, the HPMC 2208 4000 cP USP (Metolose 90 SH) was chosen as the rate controlling component of the SR formulation. This is because it is available in a special sustained/controlled release grade, where the polymers’ chain length and degrees of substitution (Methoxy and Hydroxypropyl content) are tightly controlled. Moreover, the 4000 cP grade has a short hydration time; lower swelling volume and it has acceptable hydro-gel strength, to minimise excess dissolution of the drug at the early stage of dissolution.
The last few sentences in the above extract indicate that the HPMC 2208 4000 cP was selected because of its particular suitability for use in the SR component of the formulation to be used to make the product for which marketing approval was sought.
199 The substance of the primary judge’s unchallenged findings was that the skilled addressee could produce either Formulations C or D or another formulation that was bioequivalent to either of them using the information provided in the complete specification and by performing routine experiments aimed at optimizing the selection of the high viscosity HPMC used. No witness gave evidence to suggest that the need to perform these experiments would impose an undue burden on the skilled addressee or that the non-disclosure of the particular grade and viscosity of HPMC used would inhibit the skilled addressee from obtaining the full benefit of the invention.
200 With regard to the matter of granulation, the primary judge observed at [855] that the expert witnesses acknowledged that they did not know if granule type would have had any significant impact on the dissolution profile of Formulations C or D. Referring to a MAA submitted by GSK to the TGA relating to GSK’s commercial product, the primary judge said at [859]:
[859] … The discussion of granulation optimisation again made clear that the patent applicant was at this stage concerned with optimising its particular commercial manufacturing method. It was optimising the flow and compaction procedures rather than improving the invention exemplified by Formulations C and D. Further, I agree with GSK that there was no evidence to suggest that the manufacturing method used by GSK to manufacture Formulation D on a commercial scale was anything other than conventional and routine.
201 It does not follow merely because the patent applicant uses a particular manufacturing process or a particular excipient in formulating its commercial embodiment that it will form part of the best method. The patent applicant may have adopted a particular process, or used a particular excipient, for reasons that are associated with its own particular circumstances rather than because it believes that they reflect the best method. The best method known to the patent applicant may be one that allows for the optimisation of a formulation by the skilled addressee rather than one that adheres to one specific formulation that the patent applicant seeks to commercialise.
202 We understand his Honour to have concluded that the particular granulation end points described in the MAA were not essential to the best method and were selected for reasons associated with GSK’s own manufacturing methods. They were not shown to form part of the best method known to the patent applicant at the time the complete specification was filed.
203 In the circumstances, we are not satisfied that the respondents discharged their onus of establishing that the Patent was invalid on the ground that the complete specification failed to specify the particular grade and viscosity of HPMC or granulation end points that might be used to perform the invention according to the best method known to the patent applicant. We agree with the primary judge that the best method was disclosed, albeit at a level of generality that did not include the more detailed but inessential manufacturing and production information described in the MAA applicable to the commercial embodiment.
204 In our view the respondents did not establish that GSK failed to comply with the best method requirement. The cross-appeals should be dismissed.
DISPOSITION
205 The appeals and the cross-appeals will be dismissed. We will make directions for the exchange of brief written submissions in relation to the costs of the appeals and cross-appeals.
I certify that the preceding two hundred and five (205) numbered paragraphs are a true copy of the Reasons for Judgment herein of the Honourable Justices Middleton, Nicholas and Burley. |

